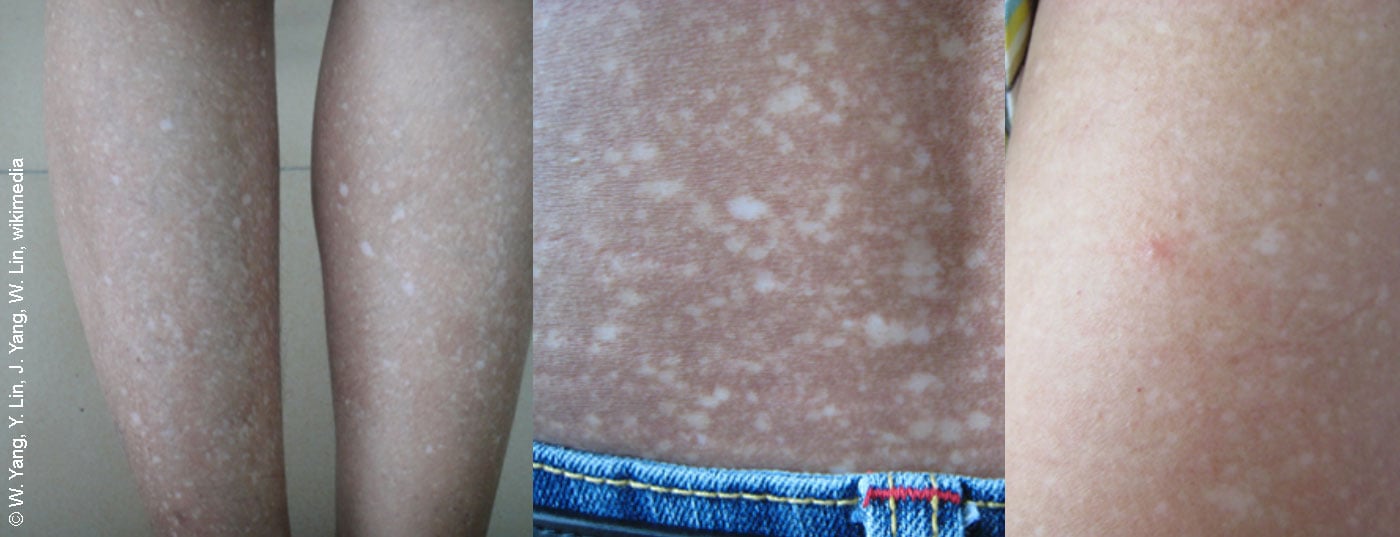
Les amyloïdoses sont une forme rare de dermatoses par dépôts, causées par un mauvais repliement des protéines. La détection précoce en dermatologie a une grande importance pour l’évolution de la maladie.
Étymologiquement, le terme amyloïdoses remonte au grec ancien ἄμυλον ámylon, qui signifie “farine de force, amidon”. Comme il s’agit biochimiquement d’une structure protéique, il s’agit d’une appellation trompeuse [1]. Les amyloïdes sont un groupe de protéines hétérogènes qui semblent similaires d’un point de vue ultrastructurel ou qui réagissent de la même manière lorsqu’elles sont colorées à l’histologie. Les amyloïdoses localisées et systémiques peuvent toutes deux être caractérisées par des symptômes cutanés. “Plus de 25 protéines sont capables de former de l’amyloïde chez les personnes en bonne santé”, a déclaré le Dr Antonio Cozzio, de la clinique de dermatologie, vénérologie et allergologie de l’hôpital cantonal de Saint-Gall, qui a fait un exposé sur ce sujet dans le cadre des Journées de formation continue en dermatologie de Zurich de cette année. Parmi les différentes protéines biochimiques capables de former l’amyloïde, 15 peuvent entraîner des maladies cliniquement significatives [2].

La taxonomie en mutation
Une détection précoce par le dermatologue constitue une base importante pour un traitement efficace et la prévention d’une détérioration supplémentaire des organes éventuellement impliqués [3]. En 2016, le système de classification de la Société des amyloïdoses a été révisé [4] : La congophilie et la biréfringence restent les principaux critères de classification. Il est également recommandé de déterminer l’identité chimique de la protéine de fibrille amyloïde déposée dans l’espace extracellulaire des tissus par l’analyse de la séquence des protéines. A l’heure actuelle, 36 protéines fibrillaires extracellulaires sont connues chez l’homme, dont 2 sont iatrogènes et 9 ont été identifiées chez l’animal. Deux nouvelles protéines fibrillaires récemment découvertes sont l’AApoCII (dérivé de l’apolipoprotéine CII) et l’AApoCIII (dérivé de l’apolipoprotéine CIII). L’amylose AApoCII et AApoCIII sont des amyloïdoses systémiques héréditaires. Deux protéines qui étaient auparavant considérées comme des inclusions intracellulaires, la tau et l’α-synucléine, sont désormais considérées comme des dépôts extracellulaires liés à la mort cellulaire et sont appelées “ATau” et “AαSyn”.
Participation cutanée hétérogène
On distingue les formes systémiques des formes cutanées limitées d’amyloïdose, chacune d’entre elles étant subdivisée en sous-types acquis et héréditaires (figure 1).
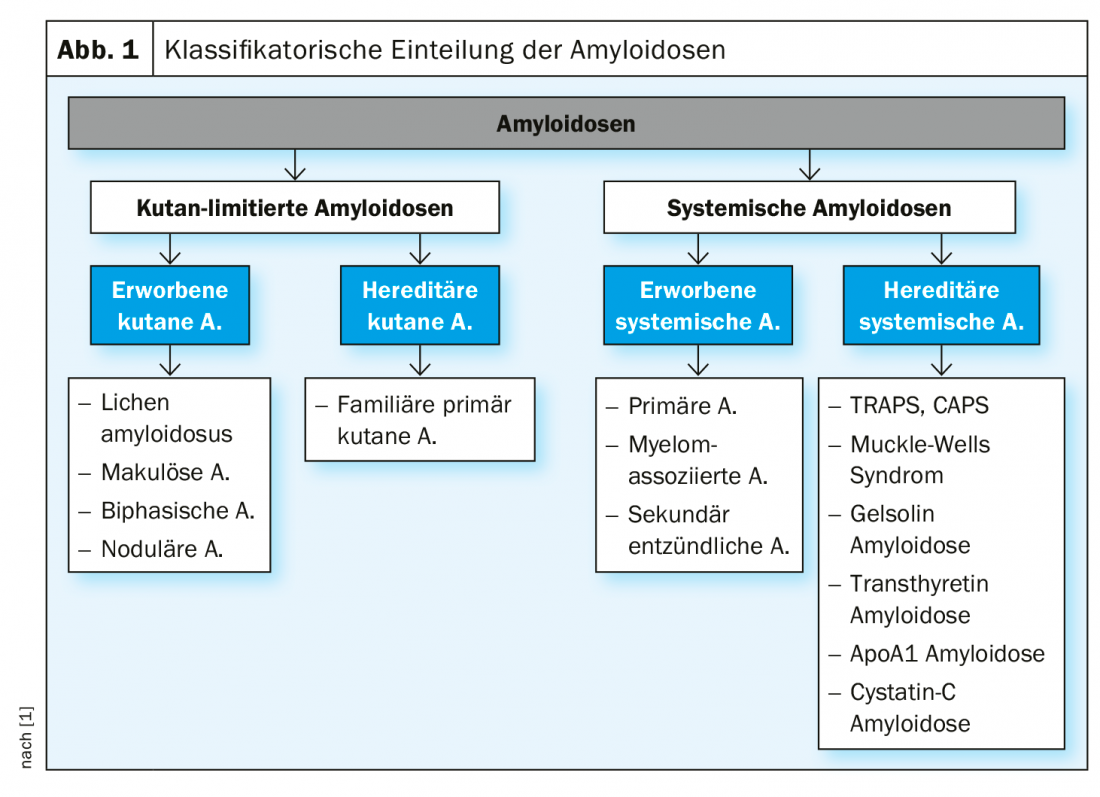
Amyloïdoses systémiques : il s’agit d’un tableau clinique hétérogène caractérisé par le dépôt de protéines pathologiquement modifiées, propres à l’organisme, dans les tissus ou les organes. Les signes cutanés caractéristiques des différentes formes d’amyloïdoses systémiques acquises sont résumés dans le tableau 1.
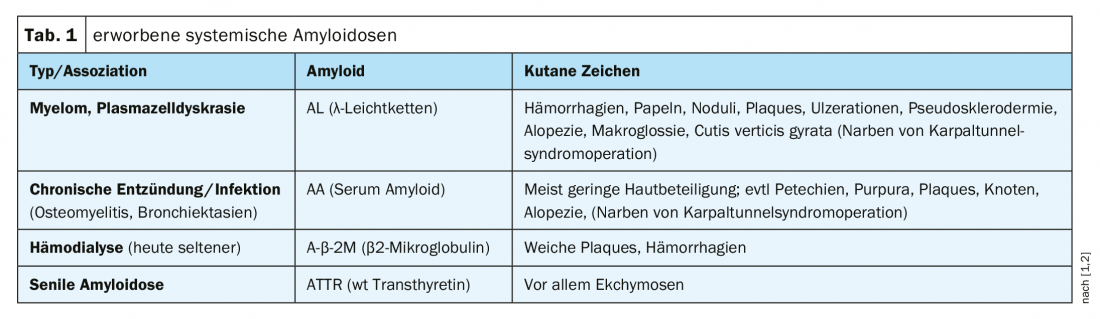
L’amylose à chaînes légères est la forme la plus fréquente et son évolution est souvent très agressive. En cas d’atteinte cardiaque ou rénale, cette forme d’amylose peut entraîner la mort en quelques mois si elle n’est pas traitée. On pense que les chaînes légères d’anticorps jouent un rôle important dans l’étiologie et conduisent aux dépôts caractéristiques dans les tissus. L’amylose systémique est fréquente chez les patients atteints de myélome multiple. Au stade précoce de l’amylose systémique primaire, les manifestations cliniques se limitent initialement à des symptômes dermatologiques, l’atteinte des organes n’intervenant que plus tard [5]. Outre la biopsie de tissus et de moelle osseuse, l’éventail des diagnostics comprend l’électrophorèse et des examens spécifiques d’organes (aperçu 1).


Amyloïdoses cutanées limitées : ce terme générique désigne des pathologies dermatologiques caractérisées histologiquement par une accumulation extracellulaire de dépôts amyloïdes dans le derme. “Les amyloïdoses maculaires et papuleuses sont généralement limitées au niveau cutané”, explique le conférencier. Si une amylose cutanée nodulaire est suspectée, il convient de déterminer s’il y a une implication cardiaque/rénale et une collaboration avec le service d’hémato-oncologie peut s’avérer nécessaire [1]. Si le purpura n’est pas clair (surtout en cas de nodules supplémentaires, de macroglossie), une biopsie avec analyse immunohistochimique est recommandée, en tenant compte des résultats histologiques antérieurs. Il convient d’exclure les diagnostics différentiels suivants : néoplasie cutanée, sarcoïdose, granulome facial, leishmaniose (aperçu 2) [1]. L’éventail des options de traitement est relativement large et doit être adapté à chaque individu (encadré).
Littérature :
- Cozzio A : Présentation de transparents : thème annuel des dermatoses de dépôt. Amyloïdoses, Dr Antonio Cozzio, 9e Journées zurichoises de formation continue en dermatologie 2019, Zurich, 26 juin 2019.
- Rauch PJ : Amyloïdoses systémiques. Forum Med Suisse 2014 ; 14(50) : 943-948.
- Bruch-Gerharz D, Ruzicka T : Amyloïdoses et hyalinoses. In : Plewig G., Ruzicka T., Kaufmann R, Hertl M (eds) Braun-Falco’s Dermatologie, Vénérologie et Allergologie 2017. Springer Reference Médecine. Berlin, Heidelberg : Springer.
- Sipe JD, et al : Amyloid fibril proteins and amyloidosis : chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloïde. The Journal of Protein Folding Disorders
- Trajber Horvat A, Trčko K, Jurčić V, Marko PB : Amyloïdose systémique primaire avec implication cutanée et cardiaque : un rapport de cas. Acta Dermatovenerologica Alpina, Pannonica, et Adriatica 2018. www.oneamyloidosisvoice.com/rcuratenew
DERMATOLOGIE PRAXIS 2019 ; 29(5) : 27-28 (publié le 10.10.19, ahead of print)