
Les troubles moteurs auto-immuns sont très rares et peuvent ressembler à des maladies neurodégénératives. C’est pourquoi il est si difficile de poser le bon diagnostic. Mais des phénotypes caractéristiques et des drapeaux rouges peuvent donner une première indication sur les anticorps sous-jacents.
Les mouvements involontaires peuvent avoir de multiples causes – et pour cette raison ne peuvent pas toujours être traités de manière causale. Certains signes d’alerte peuvent être utiles pour le diagnostic. Il s’agit notamment de l’hémichorée, pour laquelle aucune modification structurelle n’a été observée à l’IRM. L’hyponatrémie fait également partie des red flags pour lesquels il faut penser aux anticorps, a souligné le professeur Bettina Ballint, Zurich. Les anticorps anti-LGI1 peuvent se manifester par une hyponatrémie et une bradycardie prodromique. Avant que ces patients ne développent de graves déficits cognitifs, ils peuvent être traités efficacement. Les anticorps anti-LGI1 doivent également être recherchés en cas de crises dystoniques faciobrachiales. Si les troubles du mouvement se produisent également pendant le sommeil, les anticorps IgLON5 pourraient en être la cause. Ces patients présentent également un stridor important. Les anticorps IgLON5 se situent dans la zone grise entre l’auto-immunité et la neurodégénérescence. Les patients présentent à la fois une susceptibilité auto-immune et, par exemple, des tau 3R et 4R hyperphosphorylés dans l’hypothalamus et le tegmentum, sans infiltrats inflammatoires. Ces personnes ont une réponse très mitigée à l’immunothérapie.
Coup de projecteur sur la tauopathie IgLON5 dans les maladies neurodégénératives
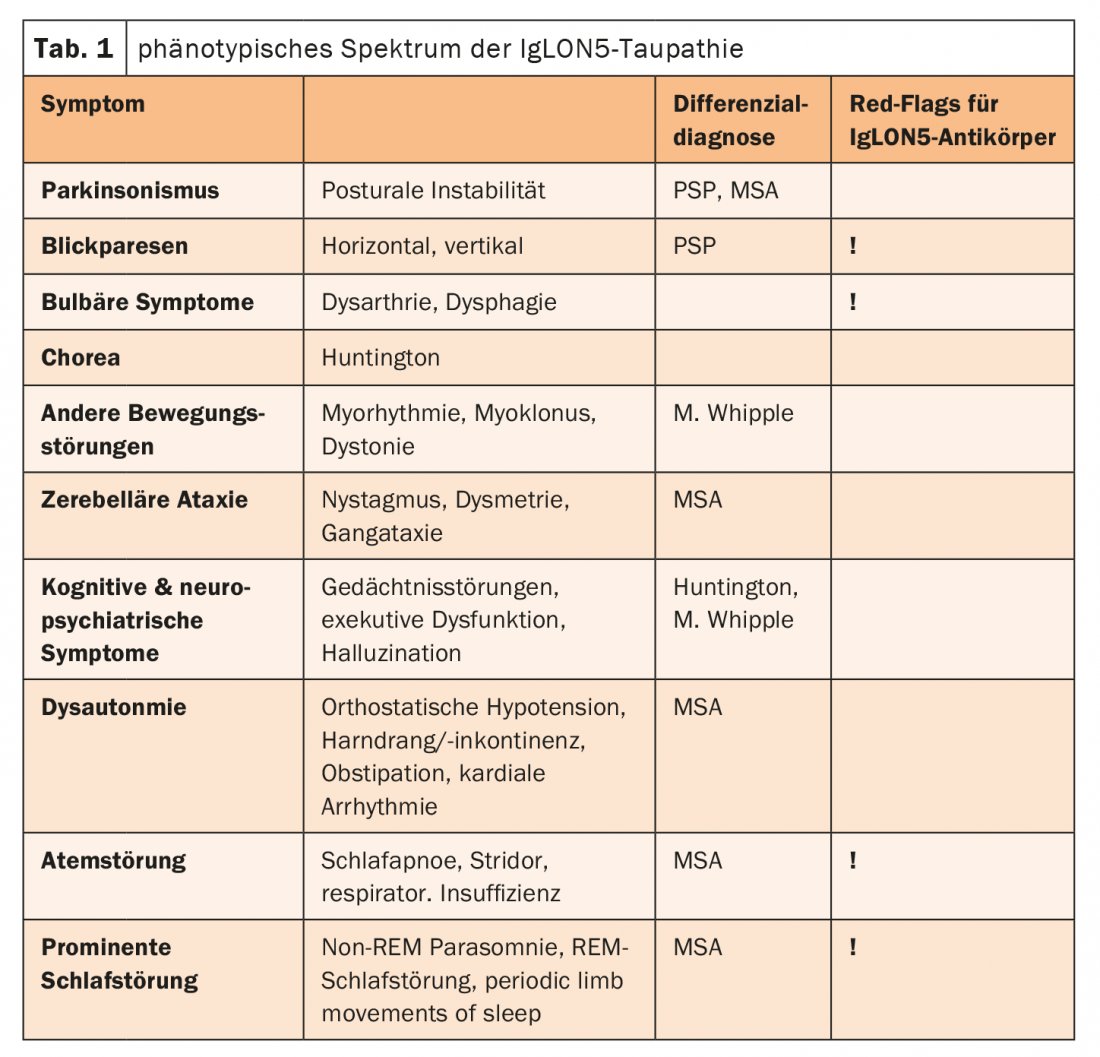
IgLON5 est une molécule d’adhésion cellulaire à la surface des neurones et joue un rôle dans le cheminement neuronal, la formation des synapses et la stabilisation des membranes. Les anticorps IgLON5-IgG1 entraînent l’internalisation et la régulation négative irréversible d’IgLON5. Les chercheurs ont donc cherché à savoir si le mécanisme d’action des anticorps, en régulant à la baisse IgLON5, perturbait l’interaction dans le cytosquelette interne et induisait ainsi l’accumulation de rosée. Cette hypothèse a été confirmée par un groupe de chercheurs de Barcelone l’année dernière, a-t-elle ajouté. Des modifications neurodégénératives avec des structures en anneau, des terminaisons précoces des dendrites ainsi que des faisceaux ont pu être mises en évidence. En outre, l’induction d’une accumulation de rosée a été démontrée. Ces effets ont été observés après trois semaines, ce qui signifie en conclusion que la tauopathie associée aux anticorps IgLON5 est une maladie à progression lente et chronique. Elle constitue un diagnostic différentiel important avec les maladies neurodégénératives primaires. Le spectre phénotypique de la tauopathie à IgLON5 est large (tableau 1) .
Les anticorps CASPR2 constituent un autre aspect important. Ils peuvent par exemple être associés à une ataxie à début tardif à l’âge adulte. Surtout si elle est associée à des douleurs et à des crises d’épilepsie. Un phénotype caractéristique est en outre la présence d’anticorps CASPR2 en cas de myoclonie de la jambe. Cela concerne surtout les hommes d’âge moyen ou tardif souffrant de douleurs neuropathiques, de fasciculations, de troubles cognitifs ou de crises d’épilepsie.
L’oratrice a résumé qu’à l’âge adulte, ce sont surtout les anticorps LGI1, IgLON5 et CASPR2 qui jouent un grand rôle. Les phénotypes les plus caractéristiques sont le FBDS, la parasomnie NREM et la myoclonie des jambes. En conséquence, il faut penser à LGI1 en cas d’hyponatrémie, à IgLON5 en cas de troubles moteurs associés au sommeil et de troubles de la déglutition/de la respiration/de la motricité oculaire, et à CASPR2 en cas de douleurs neuropathiques et de myokymie.
Source : “Seltene, aber behandelbare Bewegungsstörungen”, 07.05.2022, Prof. Dr. med. Bettina Ballint, Zurich, FomF Neurologie Update Refresher, 06-07.05.2022, Zurich et en ligne
InFo NEUROLOGIE & PSYCHIATRIE 2022 ; 20(3) : 38









