Le terme générique “hypertension pulmonaire (HTP)” englobe plusieurs types d’hypertension pulmonaire potentiellement mortels, dont les approches thérapeutiques sont très différentes et très complexes. Comme les symptômes sont souvent non spécifiques, l’établissement du diagnostic représente déjà un grand défi.
La classification clinique de l’hypertension pulmonaire (HTP) se fait actuellement en cinq groupes, comme l’a rappelé d’emblée le professeur Horst Olschewski, chef du service clinique de pneumologie, clinique universitaire de médecine interne, LKH-Universitätsklinikum Graz :
- Le groupe 1 comprend l’hypertension artérielle pulmonaire (HTAP), la forme la plus rare d’hypertension pulmonaire, qui n’est diagnostiquée qu’après avoir exclu d’autres causes sous-jacentes.
- Le groupe 2, l’hypertension pulmonaire dans les cardiopathies gauches, est de loin la forme la plus fréquente d’hypertension pulmonaire.
- Le groupe 3 comprend l’hypertension pulmonaire en cas de maladie pulmonaire et/ou d’hypoxie. Cette forme est également beaucoup plus fréquente que l’HTAP.
- Le groupe 4 comprend l’hypertension pulmonaire après une embolie pulmonaire, qui concerne jusqu’à 4% des patients ayant subi une embolie pulmonaire aiguë.
- Le groupe 5 rassemble les formes rares d’hypertension pulmonaire qui ne peuvent être clairement classées dans aucun des groupes précédents, comme l’hypertension pulmonaire liée à une sarcoïdose ou à une maladie rénale chronique.
Selon le professeur Olschewski, les groupes 2 et 3 représentent ensemble 90% de toutes les hypertensions pulmonaires, ce qui présente des dangers en termes de diagnostic différentiel.
Hétérogénéité de l’IPAH
Pour une analyse en grappes, les patients répondant aux critères de l’hypertension artérielle pulmonaire idiopathique (HAPI) ont été identifiés.
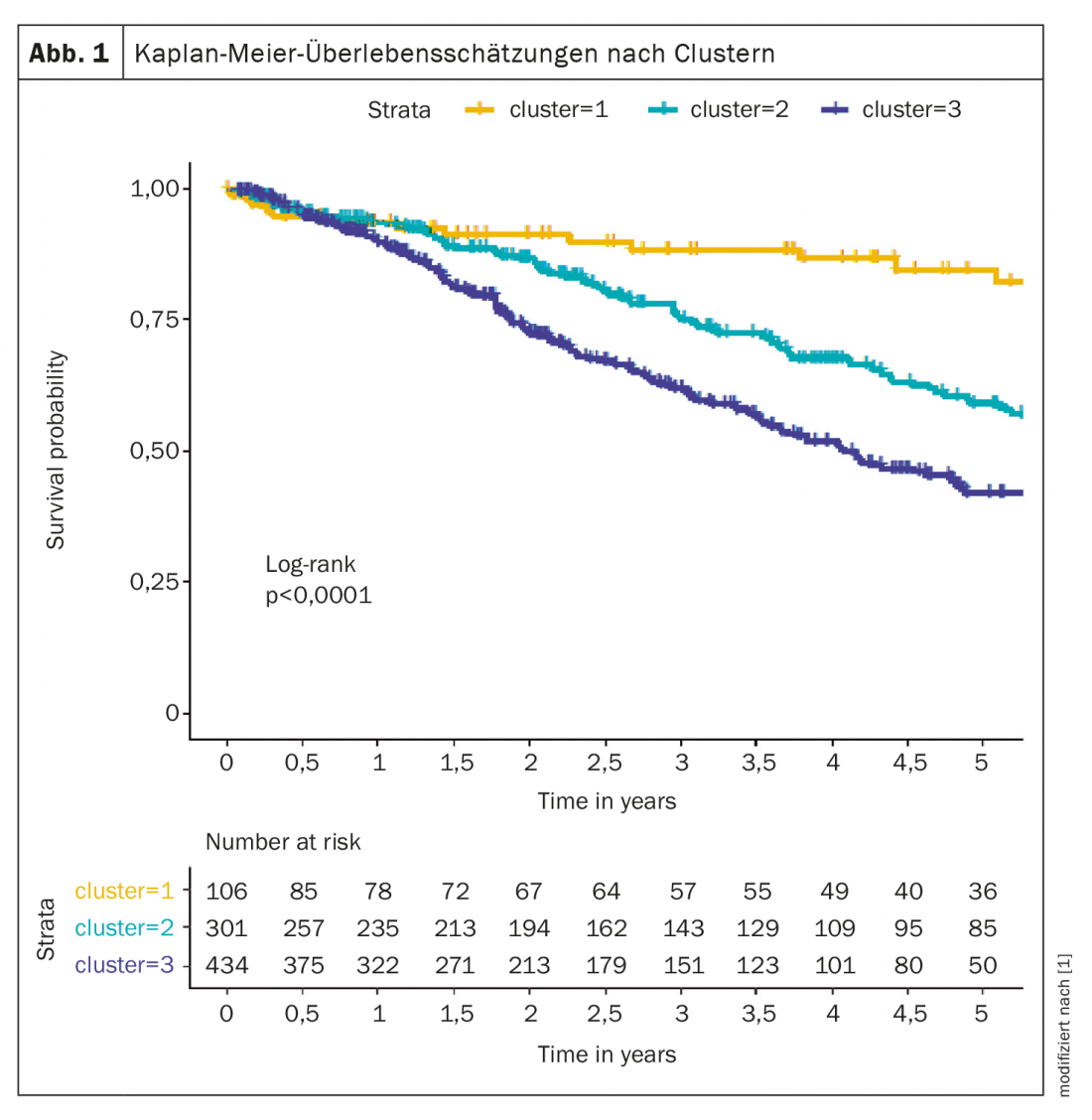
Une analyse en cluster a par exemple permis d’identifier différents phénotypes qui se distinguent par leur présentation clinique, leur réponse au traitement et leur taux de survie. Les groupes analysés ont été constitués à partir du registre COMPERA [1], trois clusters ont été définis : le cluster 1 (n=106 ; 12,6%) comprenait des patients dont l’âge moyen était de 45 ans, 76% étaient des femmes, sans comorbidités, le plus souvent jamais fumeurs, DLCO ≥45% ; le cluster 2 (n=301 ; 35,8%) comprenait des patients dont l’âge moyen était de 75 ans, 98% étaient des femmes, comorbidités fréquentes, pas d’antécédents de tabagisme, DLCO le plus souvent ≥45% ; cluster 3 (n=434 ; 51,6%) était composé de sujets d’un âge moyen de 72 ans, 72% étaient des hommes, comorbidités fréquentes, antécédents de tabagisme et DLCO en grande partie inférieure à 45%.
Les patients du cluster 1 ont mieux répondu au traitement de l’HTAP que les patients des deux autres clusters. Le taux de survie à cinq ans était de 84,6% dans le cluster 1, 59,2% dans le cluster 2 et 42,2% dans le cluster 3 (Fig. 1). Selon l’expert, ces données suggèrent que des critères sont nécessaires pour distinguer les patients IPAH atypiques des vrais patients IPAH.
Diagnostic différentiel parfois difficile
Comme les conséquences thérapeutiques dépendent en grande partie de la cause de l’hypertension pulmonaire, il est important de terminer les procédures de diagnostic et de déterminer la cause principale de l’HTP avant de prendre une décision sur les médicaments contre l’HTAP. Le World Symposia on Pulmonary Hypertension (WSPH) a établi des lignes directrices pour ces décisions importantes. Les PH du groupe 2 ou les maladies complexes du développement avec une pression post-capillaire élevée sont relativement faciles à identifier grâce à des pressions de Wedge artérielles pulmonaires élevées. L’HTP du groupe 4 peut être détectée ou exclue par des scanners pulmonaires de perfusion combinés à un scanner thoracique. Les HTAP du groupe 1 et les HTP du groupe 3 présentent des profils de maladie assez différents, mais peuvent parfois être difficiles à distinguer. La WSPH suggère qu’une hypertension pulmonaire sévère associée à une légère altération de la fonction pulmonaire (VEMS >60 et CVF >60%), à de légères anomalies parenchymateuses au scanner thoracique haute résolution et à une restriction circulatoire à l’épreuve d’effort cardiopulmonaire sont en faveur d’une HTAP du groupe 1. Ces patients sont des candidats au traitement de l’HTAP. Si le patient souffre d’une HTP du groupe 3, la seule indication possible pour un traitement de l’HTAP est une hypertension pulmonaire sévère (mPAP ≥35 mmHg ou mPAP entre 25 et 35 mmHg associée à un index cardiaque (IC) très bas <2,0 L/min/m2), qui ne peut être dérivée que de manière invasive, selon le professeur Olschewski.
Une étude s’est également penchée sur la distinction entre les patients souffrant d’HTAP (groupe 1) et ceux souffrant d’insuffisance cardiaque (groupe 2) [2]. Bien que des pressions de remplissage élevées du côté gauche et une insuffisance valvulaire mitrale fonctionnelle entraînent en premier lieu une HTP post-capillaire, les directives et les recommandations font la distinction entre l’HTP post-capillaire isolée (IpcPH) et l’HTP post-capillaire et précapillaire combinée (CpcPH). Cette dernière est définie par une résistance vasculaire pulmonaire (RVP) élevée à des unités de Wood (UB). Il est important de faire la différence entre la définition générale de l’HTP (mPAP >20 mmHg) et la définition de l’HTP précapillaire, y compris l’HTAP, pour laquelle on exige en plus une pression artérielle pulmonaire (PAWP) ≤15 mmHg et une augmentation de la résistance vasculaire pulmonaire (PVR) à ≥3 unités Wood. Selon ce document, le traitement médicamenteux ciblé d’une HTAP est indiqué lorsque la PAWP est ≤15 mmHg.
Il n’est pas non plus facile de distinguer les patients souffrant d’HTAP et de BPCO (groupe 1) des patients souffrant d’HTP en raison d’une BPCO (groupe 3). La plupart des patients atteints de BPCO avec HTP appartiennent au groupe 3. Certains patients atteints de BPCO avec HTP et une pression de remplissage du ventricule gauche élevée (HTP post-capillaire), causée par des maladies cardiovasculaires concomitantes, sont classés dans le groupe 2. Les maladies thromboemboliques chroniques peuvent également provoquer une HTP, en particulier parce que la BPCO est un facteur de risque de thromboembolie veineuse, ces patients atteints de BPCO sont généralement classés dans le groupe 4. Chez les patients présentant une obstruction très légère des voies respiratoires périphériques et une HTP précapillaire sévère avec une résistance vasculaire pulmonaire (RVP) très élevée et un faible débit cardiaque (CO), on suppose principalement que l’HTAP (groupe 1) est associée à une BPCO légère. Dans la plupart des cas, l’HTP est relativement légère chez les patients atteints de BPCO, a expliqué le pneumologue, mais dans un sous-groupe de patients atteints de BPCO, la présence de certaines caractéristiques cliniques est en faveur d’un “phénotype vasculaire pulmonaire”. Un tel phénotype serait caractérisé par une HTP précapillaire sévère avec une résistance vasculaire pulmonaire fortement accrue, une restriction modérée du flux d’air, une capacité de diffusion du monoxyde de carbone fortement réduite, une normo- ou une hypocapnie, une restriction de l’effort circulatoire et une insuffisance cardiaque droite progressive.
Une autre étude a tenté de déterminer des seuils hémodynamiques pronostiques pertinents pour l’HTP sévère dans la BPCO à l’aide d’une approche non biaisée [3]. Dans ce contexte, un PVR >5 WU s’est avéré être le plus fort prédicteur hémodynamique indépendant de la mortalité des patients atteints de BPCO. Ce seuil est le plus approprié pour identifier les patients BPCO souffrant d’une maladie vasculaire pulmonaire sévère.
Progrès dans la thérapie médicamenteuse
Les progrès récents dans le traitement médicamenteux de l’HTAP ne sont pas dus à la découverte de nouvelles voies de signalisation, mais au développement de nouvelles stratégies de thérapie combinée et à l’escalade des traitements sur la base d’une évaluation systématique de la réponse clinique. La stratégie de traitement est basée sur la sévérité du patient souffrant d’HTAP nouvellement diagnostiqué, déterminée par une approche de stratification du risque multiparamétrique. Les paramètres cliniques, d’effort, de fonction ventriculaire droite et hémodynamiques sont combinés pour définir un statut de risque faible, moyen ou élevé en fonction de la mortalité attendue à 1 an. L’algorithme de traitement actuel fournit la stratégie initiale la plus appropriée, y compris la monothérapie, la bithérapie ou la trithérapie. Une nouvelle escalade du traitement est nécessaire si le statut de faible risque n’est pas atteint lors des examens de suivi prévus. Dans la plupart des cas avancés, une transplantation pulmonaire peut être nécessaire sous traitement médical maximal (Fig. 2).
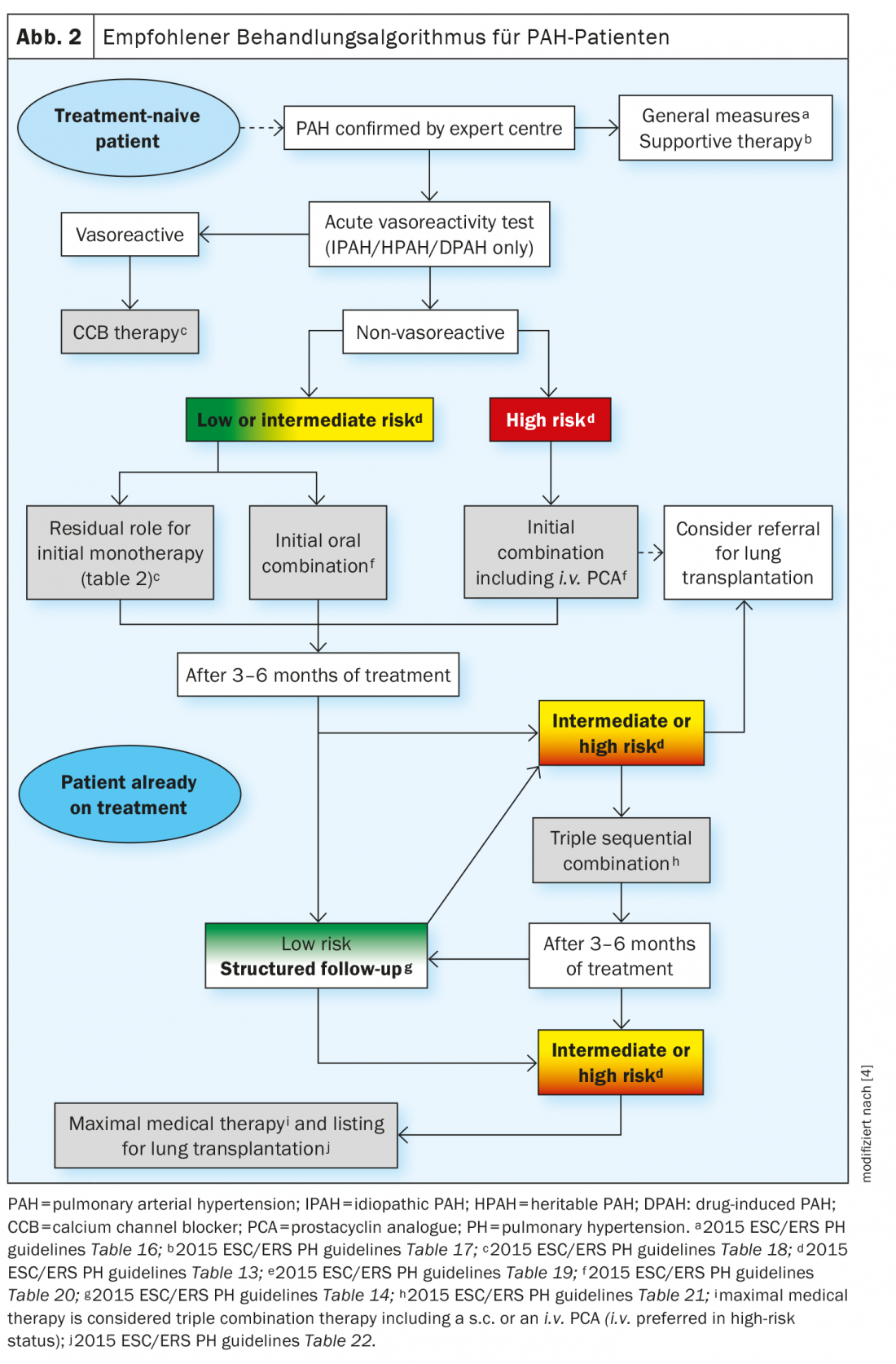
Les médicaments approuvés pour le traitement oral comprennent les inhibiteurs de la phosphodiestérase 5 (PDE5i) comme le sildénafil et le tadalafil, les antagonistes des récepteurs de l’endothéline (ERA) comme le bosentan, l’arbrisentan et le macitentan, les stimulateurs de la guanylate cyclase soluble (sGC) comme le riociguat et les agonistes des récepteurs de la prostacycline comme le sélexipag. Il est également possible de traiter l’HTAP par inhalation ou par perfusion continue.
Études prospectives sur la thérapie
Par exemple, le riociguat et les inhibiteurs de la phosphodiestérase 5 (PDE5i), qui sont autorisés dans le traitement de l’hypertension artérielle pulmonaire (HTAP), agissent par des mécanismes différents et par la même voie. L’étude REPLACE a donc cherché à déterminer si le riociguat pouvait constituer une option alternative pour les patients atteints d’HTAP qui ne répondent pas suffisamment au traitement par PDE5i. L’objectif était d’évaluer l’impact du passage au riociguat d’un traitement par PDE5i à un traitement continu par PDE5i chez des patients atteints d’HTAP présentant un risque moyen de mortalité à 1 an. Les résultats montrent que le passage du traitement par PDE5i au riociguat, qui agissent tous deux via la voie de signalisation entre la guanylate cyclase soluble dans le monoxyde d’azote et la guanosine monophosphate cyclique, peut être une option stratégique pour l’escalade thérapeutique chez les patients atteints d’HTAP présentant un risque moyen de mortalité à 1 an [5].
Le sotatercept, un nouvel antagoniste de l’activine, se lie aux activines et aux facteurs de différenciation de la croissance, tentant ainsi de rétablir l’équilibre entre les voies de signalisation favorisant et inhibant la croissance. Dans l’étude PULSAR, un traitement de 24 semaines par le sotatercept chez des patients souffrant d’hypertension artérielle pulmonaire et recevant un traitement de fond pour celle-ci a entraîné une diminution de la résistance vasculaire pulmonaire [6]. Les performances physiques (mesurées par la distance de marche de 6 minutes) et les taux de NT-proBNP se sont également améliorés sous sotatercept, selon le professeur Olschewski.
“Sensation” des États-Unis
Le professeur Olschewski a fait état d’une “sensation” aux États-Unis : Il n’y avait encore jamais eu de traitement autorisé pour la PH dans les maladies pulmonaires (groupe 3). Cela a maintenant changé – du moins aux États-Unis. C’est la première fois que le tréprostinil inhalé y est utilisé pour ce groupe de patients. L’étude INCREASE, contrôlée par placebo et d’une durée de 16 semaines, a porté sur des patients atteints de pneumopathie interstitielle et d’hypertension pulmonaire, auxquels on a administré du tréprostinil par inhalation à l’aide d’un nébuliseur à ultrasons à administration pulsée en 12 respirations maximum (72 μg au total), quatre fois par jour. Comparé au placebo, le tréprostinil inhalé a significativement amélioré les performances physiques des patients, évaluées par un test de marche de 6 minutes [7]. La substance active a déjà été approuvée par la FDA aux États-Unis, mais on ne sait pas encore si le fabricant en fera rapidement la demande auprès de l’Agence européenne des médicaments (EMA) ou s’il attendra de nouvelles études, comme l’a expliqué l’expert. “Quoi qu’il en soit, c’est une lueur d’espoir”.

L’entraînement physique standardisé est établi avec succès
L’étude randomisée contrôlée EU-TRAIN sur l’entraînement à l’exercice chez les patients atteints d’hypertension artérielle pulmonaire (HTAP) et d’hypertension pulmonaire thromboembolique chronique (HTTPC), menée dans 11 centres de 10 pays européens sur une large population de patients, a permis de constater une amélioration significative et cliniquement significative du critère d’évaluation principal 6MGT et des critères d’évaluation secondaires WHO-FC, QoL et consommation maximale d’oxygène [8]. L’étude a montré pour la première fois qu’un programme d’entraînement sûr et efficace en complément du traitement médicamenteux peut être standardisé et mis en œuvre dans différents pays avec des systèmes de santé différents.
Source : Pneumo Update 2021 : Hypertension artérielle pulmonaire, 12.11.2021
Littérature :
- Hoeper, et al : phénotypes d’hypertension artérielle pulmonaire idiopathique déterminés par analyse en grappes à partir du registre COMPERA. J Heart Lung Transplant 2020, doi : 10.1016/j.healun.2020.09.011.
- Rosenkranz, et al. : Hypertension pulmonaire en HFpEF et HFrEF : Physiopathologie, diagnostic, approches thérapeutiques. Cœur 2019, doi : 10.1007/s00059-019-4831-6.
- Zeder, et al : L’augmentation de la résistance vasculaire pulmonaire prédit la mortalité chez les patients atteints de BPCO. Eur Respir J 2021, doi : 10.1183/13993003.00944-2021.
- Galiè, et al : Risk stratification and medical therapy of pulmonary arterial hypertension. European Respiratory Journal 2019, doi : 10.1183/13993003.01889-2018.
- Hoeper, et al. : Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE) : a multicentre, open-label, randomised controlled trial. Lancet Respir Med 2021, doi : 10.1016/S2213-2600(20)30532-4.
- Humbert, et al : Sotatercept pour le traitement de l’hypertension artérielle pulmonaire. N Engl J Med 2021, doi : 10.1056/NEJMoa2024277.
- Waxman, et al : Treprostinil inhaled in Pulmonary Hypertension Due to Interstitial Lung Disease. N Engl J Med 2021, doi : 10.1056/NEJMoa2008470.
- Grüning, et al : L’exercice standardisé est faisable, sûr et efficace dans l’hypertension artérielle pulmonaire et l’hypertension pulmonaire thromboembolique chronique : résultats d’un grand essai européen multicentrique randomisé et contrôlé. Eur Heart J 2021, doi : 10.1093/eurheartj/ehaa696.
InFo PNEUMOLOGIE & ALLEROLOGIE 2022 ; 4(1) : 20-22