Les dépôts de mucine sont possibles dans toutes les couches de la peau, le lieu de dépôt n’est pas pathognomonique. Outre les mucinoses primaires, cette dermatose de dépôt se manifeste souvent comme un épiphénomène. L’étiopathogénie n’est pas totalement élucidée.
Reinhard Dummer, médecin-chef de la clinique dermatologique de l’hôpital universitaire de Zurich, et son équipe (Dr Thierry Nordmann, Dr Carole Guillet) ont fait un exposé sur les mucinoses dans le cadre du thème principal de cette année, les dermatoses de dépôt [1]. La mucine déposée est une substance constituée d’un mélange de glycosaminoglycanes également présents dans la peau saine, sous forme libre (acide hyaluronique) ou liée (protéoglycanes). Les glycosaminoglycanes sont des éléments du tissu conjonctif qui retiennent l’eau. Ils jouent un rôle important dans la consistance et la turgescence de la peau et sont produits par les fibroblastes ou les kératinocytes. La pathogenèse est soit une synthèse accrue, soit une dégradation perturbée des glycosaminoglycanes.
Challenge de diagnostic différentiel
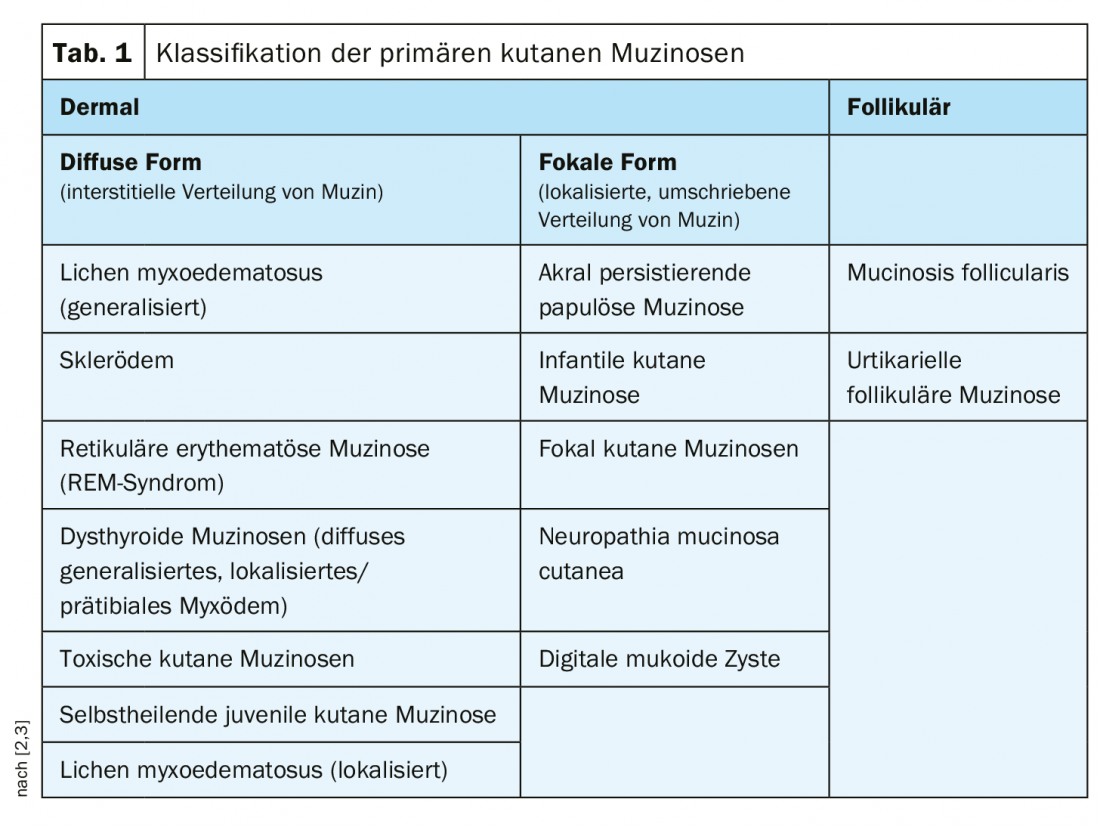
Dans les mucinoses cutanées primaires, on distingue les sous-types dermiques et folliculaires ; les sous-types dermiques sont subdivisés en une forme diffuse (distribution interstitielle de mucine) et une forme focale (distribution circonscrite localisée de mucine) (tab. 1) [2,3]. Les mucinoses primaires sont plutôt rares, l’apparition de cette dermatose de dépôt dans le cadre de processus inflammatoires et prolifératifs (phénomène réactif) est plus fréquente. Dans le lupus érythémateux et dans la dermatose granulomateuse granuloma anulare (DD Necrobiosis lipoidica), la présence accrue de mucine est un critère de diagnostic différentiel. Mais de nombreux autres processus inflammatoires peuvent également entraîner une prolifération de mucine. En outre, le tissu conjonctif peut être riche en mucine dans le stroma tumoral des tumeurs mésenchymateuses et annexielles. Une classification des mucinoses dermiques comme symptôme de différentes maladies primaires est résumée dans le tableau 2.

Syndrome du sommeil paradoxal : controverse sur l’étiopathogénie
La mucinose érythémateuse réticulée, également appelée syndrome REM, a été décrite pour la première fois en 1974 [1,3]. L’étiopathogénie du syndrome REM n’est pas encore totalement élucidée à ce jour, on discute notamment de la possibilité qu’il s’agisse d’une variante clinique du lupus érythémateux cutané [3]. Les caractéristiques cliniques typiques sont des plaques urticariennes striées et réticulées, légèrement infiltrées et rouges, situées dans la partie centrale supérieure du tronc ; la majorité des personnes atteintes sont des femmes jeunes [3]. L’exposition à la lumière du soleil peut entraîner une aggravation des symptômes [1]. En ce qui concerne les manifestations cliniques, il existe un chevauchement entre le syndrome REM et le lupus érythémateux tumide (forme évolutive du lupus érythémateux cutané) ainsi que l’infiltration lymphocytaire de type Jessner et Kanof. Jusqu’à présent, on ne sait pas exactement dans quelle mesure ces maladies constituent des entités distinctes [3]. Les fibroblastes des patients en sommeil paradoxal présentent une réponse anormale à la stimulation par l’IL-1β exogène, ce qui joue un rôle dans le métabolisme de l’acide hyaluronique potentiellement dérégulé dans le syndrome du sommeil paradoxal [1]. Les caractéristiques de diagnostic différentiel entre le syndrome REM et le lupus tumidus sont résumées dans le tableau 3.

Lichen myxoedematosus : tenir compte des diagnostics d’exclusion
Le lichen myxoedematosus est une mucinose primaire rare qui évolue de manière chronique et progressive [3]. On distingue deux sous-types : la forme localisée et la forme généralisée (tableau 4) [1]. Cette dernière est également appelée scléromyxœdème et se caractérise par des caractéristiques sclérodermiformes et papuleuses.
Les sites de prédilection du lichen myxoedematosus localisé sont le dos des mains, les faces d’extension des bras, la partie supérieure du tronc, le visage et les axilles [3]. Les caractéristiques typiques sont des papules isolées de couleur chair, rougeâtres, qui peuvent confluer et apparaître comme une lichénification et/ou un épaississement général de la peau. La distribution de mucine dans le cadre de la forme localisée peut être diffuse ou focale. La question de savoir si le lichen myxoedematosus discret, la mucinose papuleuse persistante acrale, la mucinose papuleuse auto-résorbante (forme juvénile/adulte), la mucinose infantile et la forme nodulaire doivent être considérés comme des sous-types distincts du lichen myxoedematosus localisé fait l’objet d’une controverse dans la littérature. Les symptômes classiques du sous-type généralisé (scléromyxœdème) sont des épaississements érythémateux généralisés de la peau pouvant entraîner une rigidité des mimiques et une limitation des mouvements articulaires ; dans certains cas, des anomalies cardiovasculaires ont été observées en plus des myopathies et des symptômes neurologiques [3]. Le diagnostic différentiel entre le scléromyxœdème et d’autres lésions papuleuses telles que la sclérodermie systémique et le scléroœdème doit être établi sur le plan clinique et histologique. Les résultats histologiques caractéristiques du lichen myxoedematosus local et du scléromyxœdème constituent un signe distinctif par rapport aux autres mucinoses. Elles se caractérisent notamment par un dépôt de mucine très prononcé dans le derme moyen et supérieur et souvent une prolifération des fibroblastes avec de grands noyaux en forme d’étoile ; de plus, les fibres de collagène peuvent être comprimées et proliférer (fibromuzinose) et il peut y avoir un infiltrat lymphocytaire périvasculaire de faible intensité [3]. Les glucocorticoïdes topiques et les inhibiteurs topiques de la calcineurine sont recommandés comme traitement du lichen myxoedematosus. La PUVA et les traitements au laser peuvent également être utilisés. Dans certains cas, les symptômes ont été réduits par des cytostatiques (cyclophosphamide, chlorambucil, melphalan), l’isotrétinoïne et la plasmaphérèse, ainsi que, dans des cas isolés, par des immunoglobulines intraveineuses, du melphalan à haute dose et une greffe autologue de cellules souches du sang périphérique [3]. L’orateur précise qu’en présence d’un lichen myxoedémateux localisé, la malignité et la maladie thyroïdienne sont des diagnostics d’exclusion importants [1].

Mucinose folliculaire : folliculosités proéminentes
Contrairement au syndrome REM et au lichen myxoedematosus, qui sont des sous-types dermiques, la mucinose folliculaire est caractérisée histologiquement par des dépôts de mucine dans les follicules. Il existe également un infiltrat lymphocytaire atypique et une dégénérescence de l’épithélium [1]. Le sous-type folliculaire se manifeste cliniquement par des papules et des plaques folliculaires de couleur chair à érythémateuse, disposées en foyers, avec des érosions folliculaires proéminentes (parfois avec des plaques kératosiques ressemblant à des comédons). Sur le plan étiopathogénique, une inflammation lymphocytaire initiale est discutée comme cause possible d’une accumulation secondaire de mucine. On pense qu’il existe une transition continue entre le mucinosis follicularis (plus fréquent chez les jeunes patients) et le mycosis fongoïde folliculotrope (plus fréquent chez les patients âgés) [1].
Dépôts de mucine dans le cadre de maladies multisystémiques
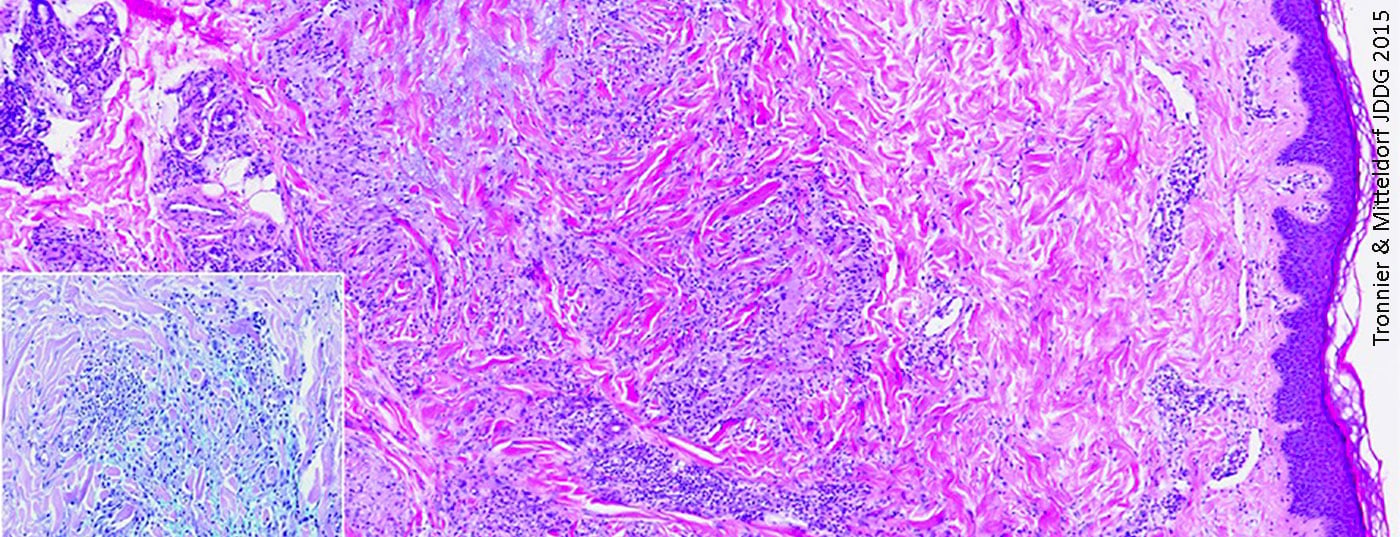
Le granulome anulaire (Fig. 1) est une maladie granulomateuse non infectieuse d’étiologie indéterminée [6], qui survient fréquemment chez des patients jeunes [7]. Les sites de prédilection des symptômes, qui se manifestent généralement sous la forme de plaques anulaires ou arciformes, sont principalement les extrémités [7]. Les dépôts de mucine dans la zone de dégénérescence du tissu conjonctif est une caractéristique histologique importante [8]. Cela est bien visible à l’histopathologie dans les colorations au bleu ou au fer colloïdal d’Alcian [8]. La plupart du temps, l’épiderme n’est pas affecté ; dans le derme, on trouve du tissu conjonctif dégénéré entouré de cellules épithélioïdes légèrement élongées et disposées en palissade [8]. Outre la forme solitaire, il existe également une forme disséminée, qui peut par exemple survenir dans le cadre d’un diabète, et qui concerne surtout les cas d’âge avancé [7].

Le lupus érythémateux est une maladie inflammatoire auto-immune dont les manifestations cliniques et l’évolution sont hétérogènes [9]. Le lupus érythémateux disséminé est une maladie multisystémique potentiellement mortelle avec une implication cutanée. L’apparition de lésions cutanées au cours de cette maladie est fréquente et peut concerner jusqu’à 70-85 % des patients [9]. Dans le lupus érythémateux aigu cutané, on distingue une forme localisée (érythème papillon) d’une variante généralisée (exanthème maculopapuleux), qui est le sous-type le plus fréquent du lupus érythémateux systémique (30-60%) [9]. Histopathologiquement, une dermatite d’interface pauvre en cellules et un infiltrat lymphocytaire périvasculaire plutôt clairsemé avec des dépôts de mucine sont des résultats typiques du lupus érythémateux aigu cutané [9].
La dermatomyosite est une autre maladie primaire avec des dépôts de mucine dermique comme caractéristique histopathologique typique [10]. Elles apparaissent dans le cadre de foyers inflammatoires au niveau de la jonction dermo-épidermique [10].
Source : ZDFT, Zurich
Littérature :
- Dummer R, Guillet C, Nordmann Th : Présentation de transparents : Thème de l’année – Dermatoses de dépôt. les mucinoses. Prof. Dr med. Reinhard Dummer & Team, 9e Journées de formation continue en dermatologie 2019, Zurich, 26 juin 2019.
- Rongioletti F, Rebora A : Mucinoses cutanées : critères microscopiques pour le diagnostic. Am J Dermatopathol 2001 ; 23 : 257-267.
- Kuhn A : Mucinoses. Dermatologie, vénérologie et allergologie de Braun-Falco 2017 ; 1-9
https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_93-1) - Schallera J, Meigelb WN : Ablagerungsdermatosen, in : Histopathologie der Haut Hrsg. : Cerroni L et al, 2ème édition 2016, Springer : Berlin Heidelberg. DOI 10.1007/978-3-662-44367-5_24-1, https://link.springer.com/content/pdf/10.1007%2F978-3-662-44367-5_24-1.pdf
- Rongioletti F, et al : Reticular erythematous mucinosis : a review of patients’ characteristics associated conditions, therapy and outcome in 25 cases. Br J Dermatol 2013 ; 169 : 1207-1211.
- Piette EW, Rosenbach M : Granulome annulaire. Pathogenesis, disease associations and triggers, and therapeutic options. JAAD 2016 ; 75 (3) : 467-479. www.jaad.org/article/S0190-9622(15)01500-5/abstract
- Houriet C, et al : Manifestations cutanées des maladies internes – 1ère partie, Clinique universitaire de dermatologie, Hôpital de l’Ile, Berne. Forum Médical Suisse 2013;13(47) : 949-953, https://boris.unibe.ch/45902/1/houriet_smf_1.pdf
- Tronnier M, Mitteldorf C : Caractéristiques histologiques des maladies granulomateuses de la peau : Partie 1 : maladies granulomateuses non infectieuses. Mini-revue. JDDG 2015 ; 13 (3) : 211-216, https://onlinelibrary.wiley.com/doi/full/10.1111/ddg.12610_suppl
- Sticherling M, Kuhn A : Lupus érythémateux. Dermatologie, vénérologie et allergologie de Braun-Falco 2017 ; 1-18, https://link.springer.com/referenceworkentry/10.1007%2F978-3-662-49546-9_54-1
- Miller ML : Diagnostic et diagnostic différentiel de la dermatomyosite et de la polymyosite chez l’adulte. UpToDate, www.uptodate.com/contents/diagnosis-and-differential-diagnosis-of-dermatomyositis-and-polymyositis-in-adults
DERMATOLOGIE PRAXIS 2019 ; 29(5) : 41-43 (publié le 10.10.19, ahead of print)