
Les symptômes psychiatriques représentent un aspect crucial de la maladie héréditaire de Huntington. Un aperçu de l’évaluation des symptômes et des options de traitement.
Les symptômes psychiatriques représentent un aspect crucial de la maladie héréditaire de Huntington. Ils apparaissent souvent même au stade prodromique et s’aggravent avec la progression de la maladie [1].
La maladie de Huntington, dont la prévalence est de 10,6 à 13,7/100 000 habitants dans le monde occidental, est une maladie polyglutaminique héréditaire autosomique dominante due à l’allongement de la répétition CAG dans le gène de Huntington sur le chromosome 4 [2,3]. Plus le nombre de répétitions est élevé, plus l’ampleur est importante et plus le temps de latence avant la première manifestation des symptômes est faible. La maladie devient cliniquement manifeste à partir d’une longueur de 39 répétitions [4]. Il s’ensuit une dégénérescence des neurones gaba- et cholinergiques dans le néostriatum et une atrophie du corps strié.
Les symptômes neuropsychiatriques peuvent être classés en quatre sous-domaines : La pulsion, l’affect, le délire et les changements de personnalité [5]. Des comportements compulsifs, des dépressions, des symptômes psychotiques, des confabulations et une évolution vers la démence peuvent survenir. Des altérations de la flexibilité cognitive ainsi que du contrôle des impulsions sont fréquentes [6].
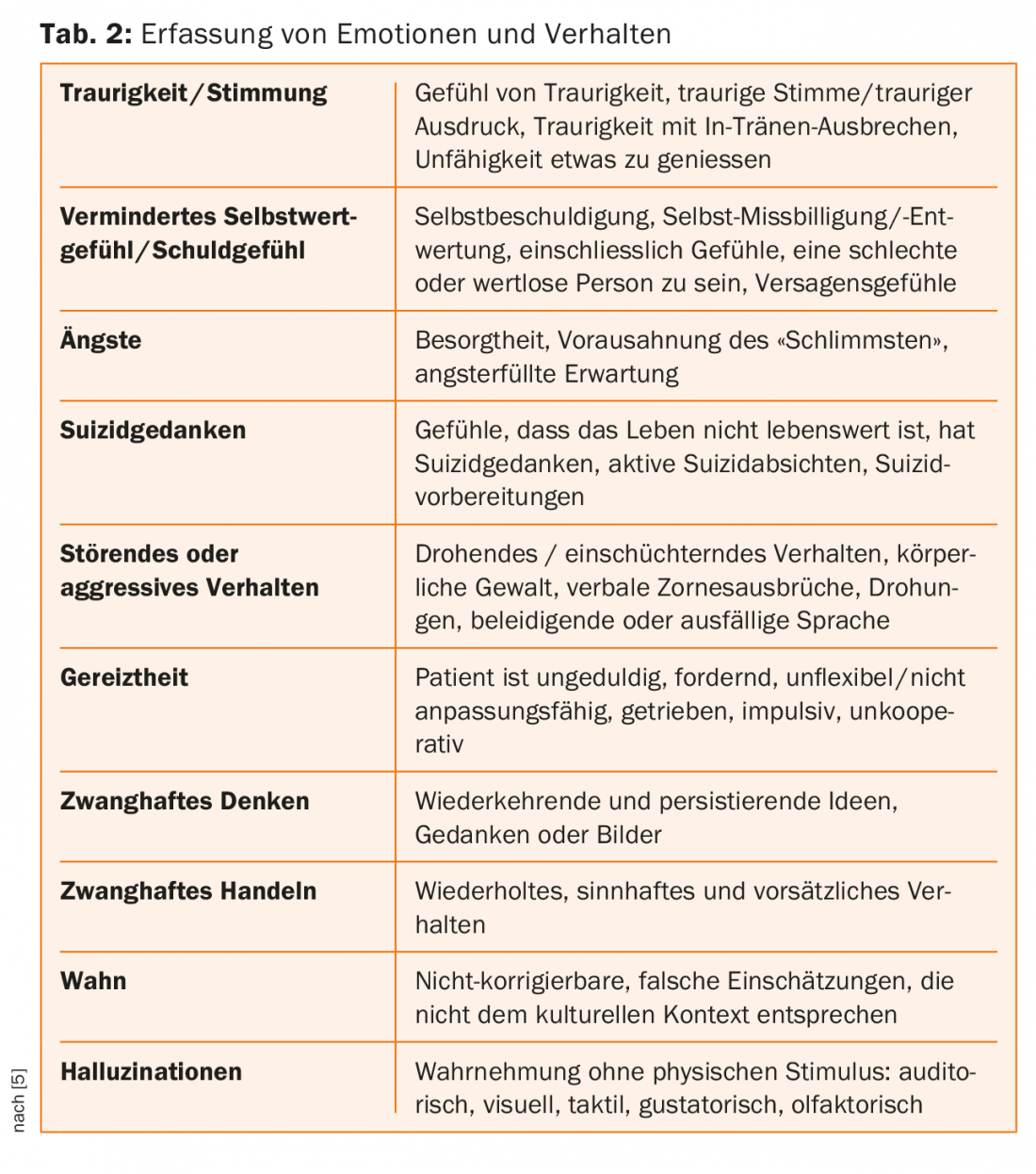
Avec l’évaluation du comportement, de la qualité de vie et du fonctionnement du patient, les symptômes psychiatriques sont évalués dans la norme d’examen “Unified Huntington’s Disease Rating Scale” (UHDRS) avec des notes sur une échelle de Likert de 0 à 4 concernant leur gravité [5] – voir Tableau 1. Outre la dépression, on observe également une irritabilité accrue au cours de la maladie ; celle-ci est souvent présente dès le stade prodromique ou le stade de manifestation précoce. La plupart du temps, l’absence de compréhension de la maladie est attribuée à la détérioration de la fonction dans le lobe frontal et à la diminution de la transmission en direction du corps striatum [7].

Cependant, ces symptômes psychiatriques peuvent également être présents dans d’autres maladies neurologiques ou psychiatriques, ce qui rend difficile le diagnostic précoce de la maladie de Huntington sur la base des symptômes psychiatriques [1].
Les symptômes moteurs typiques sont des mouvements choréiques : des contractions musculaires involontaires, arythmiques et brèves, qui se produisent dans des régions alternées du corps. À cela s’ajoutent des symptômes végétatifs tels que des troubles du sommeil, une perte de poids et une modification de la libido [6]. Dans la variante classique de la maladie, on observe très tôt – en plus des mouvements choréiques – des troubles des mouvements oculaires, des dystonies et une impotence motrice lors de l’exécution d’actions motrices simples. Les déficiences motrices réduisent l’espérance de vie des patients, notamment en raison des chutes et des pneumonies d’aspiration [2].
Anamnèse et statut
La bénéficiaire de l’AI, âgée de 42 ans, a été envoyée en thérapie résidentielle par son psychologue traitant. L’environnement familial était devenu de plus en plus conflictuel en raison du changement de caractère de la patiente, perçu de l’extérieur. Selon l’anamnèse externe, la patiente est de plus en plus irritable, agressive verbalement et de moins en moins résistante. Elle a tendance à être impulsive, à s’emporter émotionnellement et à faire des insinuations incompréhensibles aux membres de sa famille. La patiente serait devenue “plus méfiante” et se serait retirée socialement. Le matin, elle a de plus en plus de mal à se lever et manque d’énergie. Elle ne remplissait plus ses tâches ménagères et éducatives et avait parfois un “comportement bizarre”. Le mari aurait quitté le domicile en raison de la dégradation de la situation, les deux enfants communs habitant chez leur belle-sœur. La patiente elle-même a déclaré qu’elle avait l’impression que sa belle-sœur voulait lui enlever ses enfants.
Suite à un grave accident de vélo survenu il y a 18 ans, elle a été mise à la retraite AI. Pour le reste, aucun antécédent physique ou psychiatrique n’était connu et les antécédents de dépendance étaient nuls.
La patiente n’avait aucune conscience de sa maladie et peu de motivation pour le traitement. Lors de l’admission, la patiente a déclaré ne pas se rendre compte d’un trouble de la mobilité discret et involontaire. Interrogé, le mari a signalé des troubles croissants de la motricité des bras et des mains ainsi que du tronc. Aucune maladie psychiatrique ou neurologique n’est connue dans la famille.
Résultat clinique
Les résultats psychopathologiques ont montré une diminution de l’attention, de la mémoire et de la concentration. Le raisonnement formel était ordonné, la pensée resserrée. La patiente semblait méfiante, irritable et ruminante, instable sur le plan affectif, pouvant facilement dévier vers un pôle dépressif. Il y avait un délire paranoïaque concernant la belle-sœur, aucun signe d’hallucinations sensorielles ou de troubles du moi. En dehors d’une agitation motrice, la patiente ne présentait aucun signe psychovégétatif. Il n’y a pas eu d’indication d’une mise en danger aiguë de soi ou d’autrui. Sur le plan neurologique, outre les mouvements chorégraphiques discrets, l’état résiduel après un accident de la route et une reconstruction plastique (plastie du latissimus dorsi) de la jambe était impressionnant, avec une parésie et des troubles de la sensibilité dus à une paralysie post-traumatique du péroné. De légers troubles de la motricité fine et de la coordination ont été observés, ainsi qu’une démarche incertaine et asymétrique avec une boiterie raidie. Les sensations de pression, de température et de douleur au niveau de la jambe gauche ont été signalées comme étant accrues.
Diagnostic et évolution clinique
Un peu plus d’un mois après son admission in domo, il a été présenté à la clinique neurologique de l’hôpital cantonal de Lucerne sur la base d’un diagnostic de suspicion de maladie de Huntington. Là, l’examen physique a révélé une apraxie du regard, un langage intermittent de type staccato et des dyskinésies de la bouche, de la mâchoire et des yeux, ainsi qu’une myoclonie des coins de la bouche et des dyskinésies myocloniformes des doigts des deux côtés. En outre, on a constaté une résistance motrice de la langue et des mains (“milkmaids grip”), une dysdiadochocinésie et des mouvements choréiques intermittents, ainsi qu’une augmentation des réflexes musculaires propres de tous les membres et un réflexe cloniforme du tendon d’Achille des deux côtés. Une ataxie de la station debout et de la marche s’est manifestée, avec une augmentation des dyskinésies.
Les examens réalisés, à savoir l’EEG, l’hyperventilation et la photostimulation, ainsi que l’échographie duplex des vaisseaux cérébraux, n’ont pas révélé de résultats pathologiques probants. Une IRMc réalisée a mis en évidence une atrophie caudataire bilatérale sans troubles de la signalisation et une légère atrophie putaminale.
La suspicion de maladie de Huntington s’est ainsi renforcée, et le diagnostic différentiel a porté sur une dyskinésie tardive due à d’anciens anesthésiques et sur une maladie de Wilson.
Sur le plan thérapeutique, l’hôpital psychiatrique a mis en place de la quétiapine 300 mg pour traiter les symptômes affectifs et délirants, et la patiente a également été intégrée dans des thérapies orientées vers l’action.
Un peu plus de deux mois après son admission, la patiente a quitté l’établissement à sa demande et après consultation du service de neurologie, tout en continuant à prendre de la quétiapine retard 300 mg. Le jour de sa sortie, un examen de génétique moléculaire a été effectué à l’hôpital cantonal de Lucerne, qui a révélé une expansion pathologique des CAG avec 46 répétitions des CAG, ce qui a permis de poser le diagnostic de certitude de la maladie de Huntington.
Malgré des symptômes psychiques pertinents pour le traitement, un traitement antidyskinétique n’était pas encore nécessaire en raison de la légèreté des symptômes moteurs à la sortie. En plus des conseils génétiques et psychologiques, un service social et des soins de proximité ont été recommandés en post-hospitalisation.
Discussion
La maladie de Huntington est une maladie neuropsychiatrique facile à diagnostiquer au stade tardif grâce aux mouvements choréiques (voir Tab. 1-3 pour enregistrer les troubles du comportement). Cependant, des anomalies neuropsychiatriques non spécifiques apparaissent souvent des années avant la première manifestation des symptômes moteurs et affectent fortement la vie des patients. Il est d’autant plus important d’inclure une telle “maladie orpheline” dans le diagnostic différentiel des patients présentant des troubles psychiatriques, afin de pouvoir les accompagner et les soigner de manière adéquate [8]. Dans notre cas, l’irritabilité paranoïaque et méfiante, le manque de contrôle des impulsions et l’humeur dépressive étaient les principaux facteurs, en plus des mouvements chorégraphiques initiaux.
Jusqu’à présent, il n’existe aucune possibilité de traitement causal, mais de nombreuses recherches sont en cours pour déterminer la meilleure façon d’influencer la progression de la maladie [9]. Les premières études médicales en laboratoire indiquent des possibilités de traitement dans un avenir proche [10]. Les traitements curatifs possibles sont les guides génétiques, les biomarqueurs ou les greffes de cellules souches, mais ils ne sont pas encore utilisables et le diagnostic est donc toujours considéré comme incurable [9].
Après l’annonce du diagnostic, le patient a besoin de soins et de conseils complets. Outre les thérapies fonctionnelles (ergothérapie, physiothérapie, orthophonie), le début d’un traitement médicamenteux est indiqué en présence de symptômes psychiques nécessitant un traitement – comme dans le cas décrit, par exemple avec des médicaments stabilisant l’humeur et à effet antipsychotique tels que les neuroleptiques atypiques. Si nécessaire, il est possible de traiter les hyperkinésie gênantes avec de la tétrabénazine ou du tiapride et les symptômes parkinsoniens avec de la L-dopa. Le conseil génétique ainsi que le soutien social et psychothérapeutique des patients et des familles concernés sont d’une grande importance.

Messages Take-Home
- La chorée de Huntington peut provoquer des symptômes neuropsychiatriques invalidants dès la phase prodromique ou à un stade précoce.
- Les symptômes neuropsychiatriques proviennent de quatre sous-domaines : pulsion, affect, délire et altération de la personnalité.
- Le patient n’a souvent pas conscience de sa maladie, même si ses proches et son entourage souffrent.
- Une évaluation des symptômes peut être effectuée, par exemple, par le biais de la saisie des troubles du comportement dans l’échelle “Unified Huntington’s Disease Rating Scale” (UHDRS).
- Les antipsychotiques atypiques et l’accompagnement psychothérapeutique peuvent être utilisés dans le traitement des symptômes neuropsychiatriques.
Littérature :
- Epping EA, Kim JI, et al : Longitudinal Psychiatric Symptoms in Prodromal Huntington’s Disease : A Decade of Data. Am J Psychiatry. 2016 ; 173(2) : 184-192.
- Pringsheim T, Wiltshire K, et al. : The incidence and prevalence of Huntington’s disease : a systematic review and meta-analysis. Mov Disord. 2012 ; 27(9) : 1083-1091.
- McColgan P, Tabrizi SJ : Huntington’s disease : a clinical review. Eur J Neurol. 2018 ; 25(1) : 24-34.
- Paulson HL, Albin RL : Maladie de Huntington : caractéristiques cliniques et voies d’accès au traitement. In : Neurobiologie de la maladie de Huntington : Applications à la découverte de médicaments (eds Lo DC, Hughes RE) : 2011.
- Kieburtz K : Unified Huntington’s Disease Rating Scale : Reliability and-Consistency. Troubles du mouvement. 1996 II(2) : 136-142.
- Duff K, Paulsen JS, et al : Predict HDIotHSG. Symptômes psychiatriques dans la maladie de Huntington avant le diagnostic : l’étude predict-HD. Biol Psychiatry. 2007 ; 62(12) : 1341-1346.
- Hoth KF, Paulsen JS, et al : Les patients atteints de la maladie de Huntington ont une conscience altérée de leurs capacités cognitives, émotionnelles et fonctionnelles. J Clin Exp Neuropsychol. 2007 ; 29(4) : 365-376.
- Schiefer J, Werner C, et al : Diagnostic clinique et prise en charge dans la maladie de Huntington précoce : une revue. Maladie neurologique et neuromusculaire dégénérative. 2015 ; (5) : 37-50.
- Lo DC, Hughes RE, editors. Neurobiologie de la maladie de Huntington : Applications à la découverte de médicaments. CRC Press/Taylor & Francis, 2011.
- Chen GL, Ma Q, et al : Modulation of nuclear REST by alternative splicing : a potential therapeutic target for Huntington’s disease. J Cell Mol Med. 2017 ; 21(11) : 2974-2984.
Remerciements : Pour leur soutien clinique et leur collaboration, nous remercions vivement le professeur Bohlhalter et le docteur Stephan Mittas du service de neurologie de Lucerne, dipl. med. Berennen-Dietrich de l’Institut Rötgen de Lucerne et le Dr. med. Roland Spiegel de Genetica Zurich.
InFo NEUROLOGIE & PSYCHIATRIE 2018 ; 16(1) : 24-27









