Il trattamento dell’ipertensione arteriosa polmonare (PAH) comprende misure non specifiche (terapia di base) e farmaci specifici (vasodilatatori polmonari selettivi). Una terapia di base ben sviluppata, che includa l’anticoagulazione orale, il controllo del ritmo, le misure di riabilitazione e il supporto psicosociale per i sintomi depressivi, è essenziale. I vasodilatatori specifici riducono la morbilità e la mortalità della PAH. Attualmente sono disponibili diverse sostanze che influenzano una delle tre vie di segnalazione degli IPA: Cascata di endotelina, NO o prostaciclina. Le sostanze che influenzano la stessa via di segnalazione (ad esempio, riociguat e sildenafil o macitentan e bosentan) non devono essere combinate.
L’ipertensione arteriosa polmonare (PAH) è una malattia della vascolarizzazione polmonare periferica che, se non trattata, porta ad un aumento progressivo della resistenza vascolare polmonare e ad una riduzione delle dimensioni della circolazione polmonare. La prognosi è determinata dalla capacità del ventricolo destro di tollerare questo aumento del postcarico.
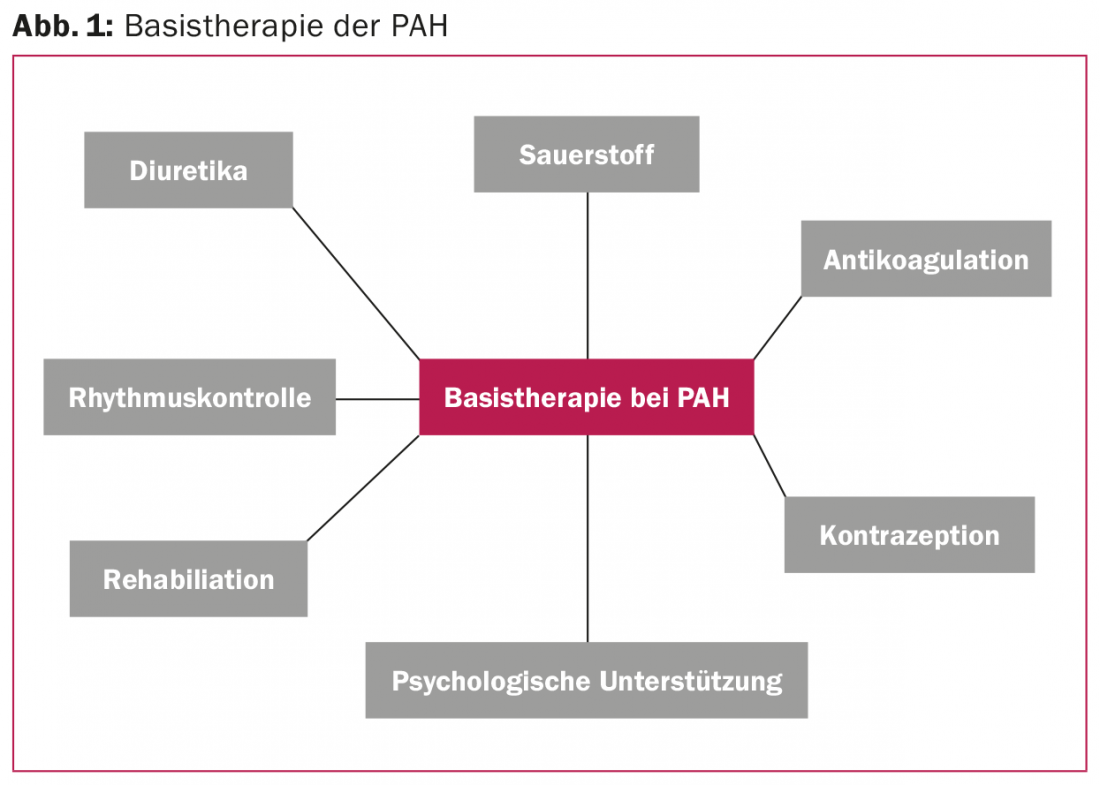
Circa 30 anni fa, la sopravvivenza mediana dopo la diagnosi di “ipertensione polmonare primaria” era di 2,8 anni [1]. Dai primi approcci terapeutici con i calcio antagonisti e l’anticoagulazione orale, le opzioni terapeutiche si sono moltiplicate. Mentre all’inizio ci si concentrava su misure non specifiche, come il trattamento dell’insufficienza cardiaca destra con diuretici e digossina, nel frattempo sono stati sviluppati farmaci che influenzano selettivamente diverse vie di segnalazione della vasocostrizione polmonare e inibiscono la progressione della malattia. Dieci sostanze sono attualmente approvate per il trattamento della PAH [2]. Oltre a questa vasodilatazione polmonare selettiva, tuttavia, la terapia di base adattata individualmente non ha perso la sua importanza e dovrebbe integrare qualsiasi terapia specifica per la PAH (fig. 1).
Vasodilatatori polmonari selettivi
La disfunzione endoteliale e il conseguente squilibrio tra vasocostrittori e vasodilatatori è un aspetto importante nella patogenesi della PAH. La disfunzione endoteliale influisce sul rimodellamento vascolare della circolazione polmonare, nonché sull’emostasi e sulla coagulazione, che a loro volta contribuiscono alla progressione della PAH. I farmaci per la PAH utilizzati oggi hanno come bersaglio una delle tre vie di segnalazione conosciute che influenzano la pressione polmonare: Prostaciclina (PGI2) [3], Azoto (NO) [4] e la via dell’Endotelina-1 [5] (Fig. 2).
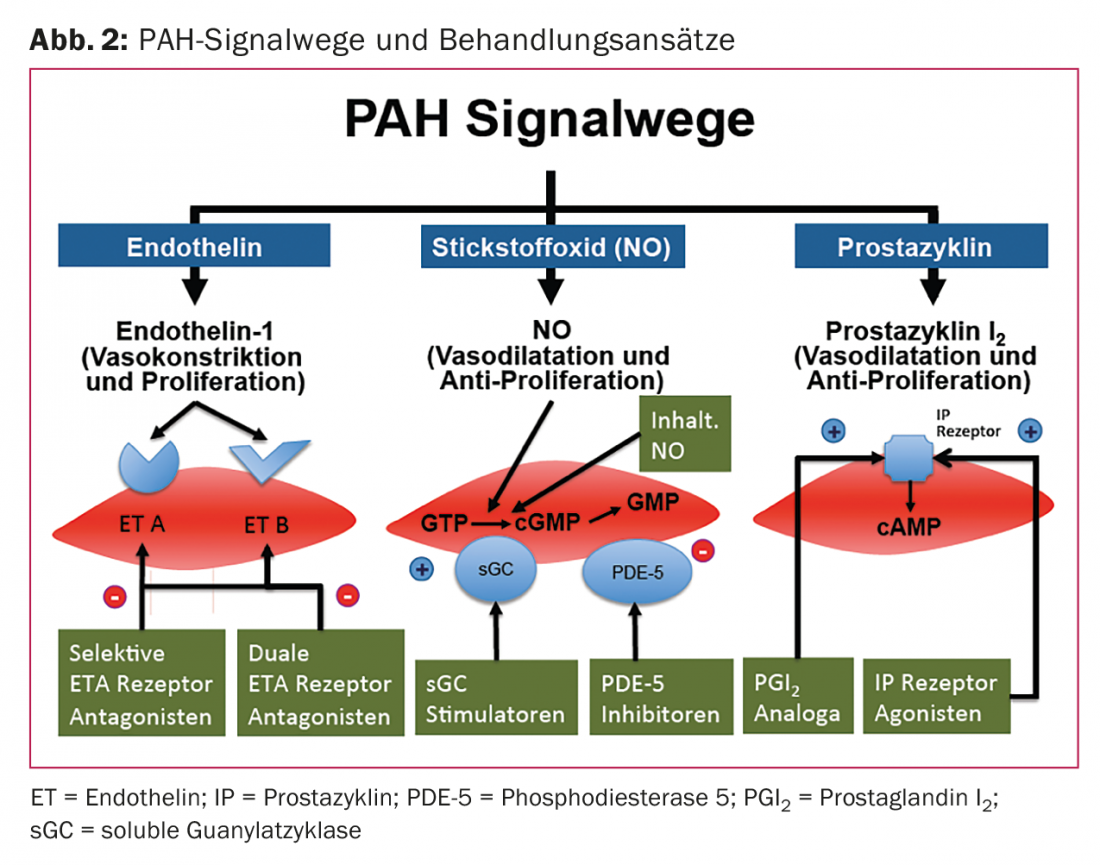
La PAH è caratterizzata da un aumento dei vasocostrittori (ad esempio, l’endotelina-1) e da una riduzione dei vasodilatatori (ad esempio, PGI2 e NO).
Dal 2004 sono disponibili vasodilatatori polmonari di tutte e tre le vie. Negli ultimi anni, sono stati testati farmaci che sono ulteriori sviluppi di molecole esistenti (ad esempio, macitentan), mirano a nuovi punti della cascata di segnalazione (ad esempio, riociguat) o contengono una molecola nuova che può legarsi a recettori identici a quelli delle sostanze esistenti (ad esempio, selexipag). Si spera che questi nuovi agenti abbiano proprietà farmacocinetiche migliorate che semplifichino la somministrazione, aumentino l’efficacia e/o siano associati a un profilo di effetti collaterali inferiore.
Macitentan
Macitentan (Opsumit®) è il principio attivo più recente del gruppo degli antagonisti del recettore dell’endotelina. Altri antagonisti del recettore dell’endotelina approvati in Svizzera sono bosentan (Tracleer®) e ambrisentan (Volibris®). L’endotelina-1 ha un pronunciato potenziale vasocostrittivo ed effetti stimolanti sui leucociti. Il legame al recettore dell’endotelina accoppiato a proteine G isoforme A (ETA) e B (ETB) delle cellule muscolari lisce media la vasocostrizione delle arterie polmonari. Come il bosentan, il macitentan agisce attraverso un blocco del doppio recettore. Grazie ai metaboliti attivi più lunghi e alla dissociazione ritardata del recettore, si ipotizza un migliore effetto di abbassamento della pressione per il macitentan [6].
L’efficacia del macitentan è stata studiata in uno studio di fase III (studio SERAPHIN) su 742 pazienti [7]. Hanno ricevuto placebo, macitentan 3 mg/die o macitentan 10 mg/die in un rapporto 1:1:1. Inoltre, potrebbe essere utilizzata una terapia con prostanoidi o inibitori della fosfodiesterasi. SERAPHIN è il più grande studio a lungo termine, in doppio cieco, randomizzato, controllato con placebo, condotto su pazienti con PAH e presenta un approccio innovativo come studio di esito guidato dagli eventi. Sulla base di questi dati, è stato dimostrato che macitentan riduce l’endpoint combinato di morbilità e/o mortalità del 45% nei pazienti con PAH rispetto al placebo.
La Figura 3 mostra i grafici Kaplan-Meier dell’effetto a lungo termine in termini di morbilità e mortalità nei pazienti con e senza terapia combinata con altri vasodilatatori polmonari.
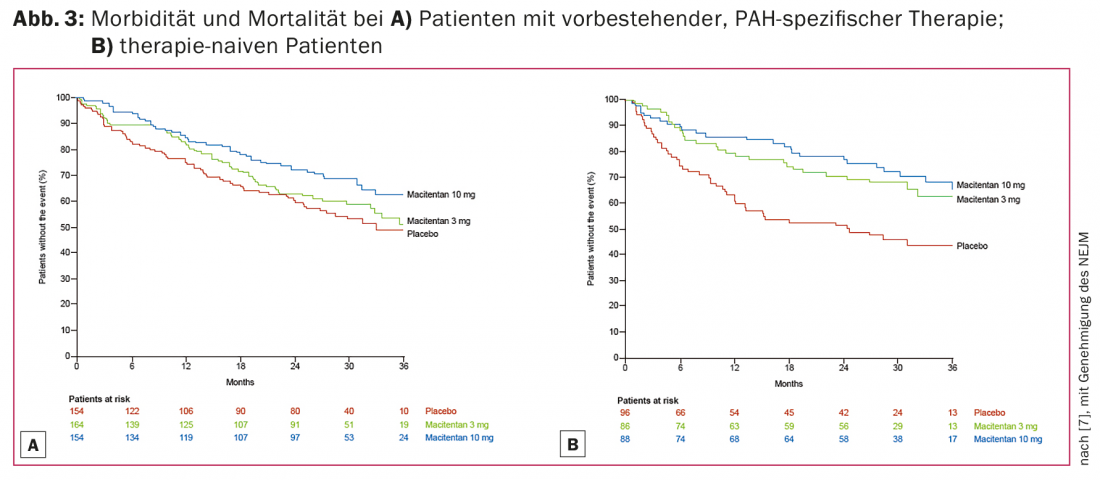
Sulla base di questi dati di studio, macitentan 10 mg al giorno è stato approvato da febbraio 2014 per il trattamento della PAH con una classe funzionale II-III secondo la definizione dell’OMS. Rispetto al bosentan, già affermato, induce meno interazioni farmacologiche e ha meno effetti collaterali; in particolare, non è necessario controllare mensilmente le transaminasi. Gli effetti collaterali più comuni sono nasofaringite, anemia e cefalea.
Il macitentan viene scomposto principalmente nel fegato attraverso il citocromo P450 3A4 (CYP3A4). Bisogna quindi notare che gli induttori del CYP3A4 (erba di San Giovanni, anticonvulsivanti, rifampicina, ecc.) e gli inibitori del CYP3A4 (succo di pompelmo, antibiotici macrolidi, metronidazolo, antimicotici, HAART, ecc.
Riociguat
Riociguat appartiene a una nuova classe di farmaci per la terapia della PAH. Come gli inibitori convenzionali della fosfodiesterasi-5 (PDE-5), funziona attraverso la via di segnalazione vasodilatatrice dell’ossido nitrico (NO). Il Riociguat stimola la guanilato ciclasi indipendentemente dall’NO, determinando un aumento del guanosina monofosfato ciclico intracellulare (cGMP). Il cGMP a sua volta influenza la concentrazione di calcio e quindi la contrattilità delle cellule muscolari lisce delle arterie polmonari. Gli inibitori della PDE-5 convenzionali inibiscono la degradazione del cGMP. Il loro effetto richiede quindi una produzione basale di cGMP mediata da NO. Poiché la PAH è associata a una ridotta produzione endogena di NO, questo potrebbe limitare il profilo di efficacia degli inibitori della PDE-5. Questo svantaggio teorico viene aggirato con riociguat.
Ampi studi controllati con placebo hanno dimostrato l’efficacia di riociguat nel migliorare la distanza di cammino nel test del cammino di 6 minuti per i pazienti con PAH (studio PATENT) [8] e per i pazienti con ipertensione polmonare cronica tromboembolica non operabile o persistente/recidivante dopo tromboendarterectomia (studio CHEST) [9]. Entrambi gli studi di fase III hanno confermato un buon profilo di sicurezza, con emorragie polmonari verificatesi in pochi casi.
Il dosaggio di riociguat deve essere determinato individualmente; la dose massima è di 2,5 mg tre volte al giorno. Gli effetti collaterali più comuni sono ipotensione, cefalea, vertigini e disturbi gastrointestinali. Dispepsia. La combinazione con gli inibitori della PDE-5 o con i preparati di NO è controindicata, e le combinazioni con gli agenti antiaggreganti devono essere ben valutate caso per caso.
Selexipag
Selexipag è il primo agonista del recettore della prostaglandina I2 disponibile in compresse che non consiste in una molecola di prostaciclina. Finora si potevano utilizzare solo gli analoghi della prostaciclina, che dovevano essere somministrati continuamente per via endovenosa o sottocutanea o inalati ogni tre ore, a causa di un’emivita molto breve.
La via di segnalazione della prostaciclina ha un ruolo particolare nel trattamento della PAH, in quanto solo l’epoprostenolo (analogo della prostaglandina I2 per somministrazione endovenosa continua) ha dimostrato di ridurre direttamente la mortalità in uno studio controllato con placebo [10]. Le prostacicline in somministrazione continua sono il trattamento di scelta per la PAH scompensata.
L’effetto di selexipag è stato studiato per la prima volta in un piccolo studio di fase II su 43 pazienti già in terapia per la PAH con antagonisti dei recettori dell’endotelina o inibitori della PDE-5 (o entrambi) [11]. Con selexipag, è stata ottenuta una riduzione della resistenza polmonare di >30%, indipendentemente dal fatto che i pazienti fossero già stati pretrattati o meno.
Nello studio Griphon di fase III, con oltre 1100 pazienti e un endpoint clinico combinato (mortalità e morbilità), il beneficio di selexipag si è dimostrato ancora una volta indipendente dal sesso, dai farmaci preesistenti, dalla gravità della compromissione delle prestazioni, dall’età e dalla causa della PAH. L’endpoint primario è stato raggiunto dal 42% dei pazienti con placebo contro il 27% dei pazienti con selexipag dopo una media di 70 settimane [12].
Partendo dal presupposto che la densità dei recettori della prostaciclina varia da paziente a paziente e che quindi sono necessarie dosi diverse per ottenere il blocco massimo, è stato previsto un up-titration della dose di selexipag (400-3200 µg/d). Se si sono verificati effetti collaterali (mal di testa, diarrea, nausea o dolore alla mascella), è stata scelta la dose successiva più bassa come dose di mantenimento. Poiché non è stata osservata alcuna differenza tra i pazienti con dosi elevate e basse nell’analisi dell’endpoint, si presume che questo concetto sia dimostrato (Fig. 4).
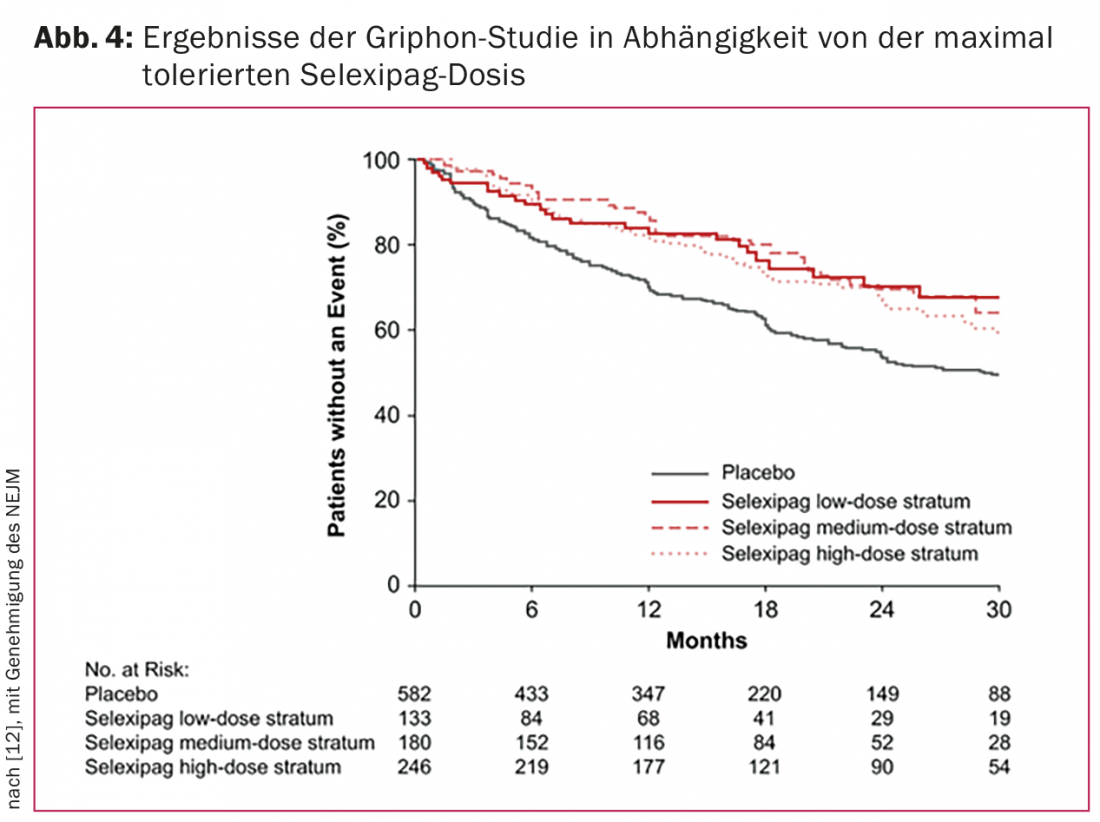
Selexipag sarà approvato in Svizzera a partire dalla metà del 2016. Simile al farmaco riociguat, sarà un altro farmaco per la PAH che non viene prescritto in una dose fissa, ma deve essere titolato individualmente. Come tutte le altre sostanze, deve essere prescritta solo da medici esperti nel trattamento della PAH, idealmente attraverso centri PAH affermati.
Letteratura:
- Rich S, et al: Ipertensione polmonare primaria. Uno studio prospettico nazionale. Ann Intern Med 1987; 107(2): 216-223.
- Humbert M, et al: Progressi negli interventi terapeutici per i pazienti con ipertensione arteriosa polmonare. Circolazione 2014; 130(24): 2189-2208.
- Giaid A, Saleh D: Ridotta espressione dell’ossido nitrico sintasi endoteliale nei polmoni dei pazienti con ipertensione polmonare. N Engl J Med 1995; 333(4): 214-221.
- Christman BW, et al: Uno squilibrio tra l’escrezione dei metaboliti del trombossano e della prostaciclina nell’ipertensione polmonare. N Engl J Med 1992; 327(2): 70-75.
- Giaid A, et al: Espressione dell’endotelina-1 nei polmoni dei pazienti con ipertensione polmonare. N Engl J Med 1993; 328(24): 1732-1739.
- Iglarz M, et al.: Confronto dell’attività farmacologica di macitentan e bosentan in modelli preclinici di ipertensione sistemica e polmonare. Life Sci 2014; 118(2): 333-339.
- Pulido T, et al: Macitentan e morbilità e mortalità nell’ipertensione arteriosa polmonare. N Engl J Med 2013; 369(9): 809-818.
- Ghofrani HA, et al: Riociguat per il trattamento dell’ipertensione arteriosa polmonare. N Engl J Med 2013; 369(4): 330-340.
- Ghofrani HA, et al: Riociguat per il trattamento dell’ipertensione polmonare cronica tromboembolica. N Engl J Med 2013; 369(4): 319-329.
- Barst RJ, et al: Un confronto tra l’epoprostenolo continuo per via endovenosa (prostaciclina) e la terapia convenzionale per l’ipertensione polmonare primaria. Il Gruppo di Studio sull’Ipertensione Polmonare Primaria. N Engl J Med 1996; 334(5): 296-302.
- Simonneau G, et al: Selexipag: un agonista orale selettivo del recettore della prostaciclina per il trattamento dell’ipertensione arteriosa polmonare. Eur Respir J 2012; 40(4): 874-880.
- Sitbon O, et al: Selexipag per il trattamento dell’ipertensione arteriosa polmonare. N Engl J Med 2015; 373(26): 2522-2533.
CARDIOVASC 2016; 15(2): 6-10