
Questo termine generico copre le malattie metaboliche molecolari geneticamente estremamente eterogenee e prevalentemente ereditarie della biosintesi ematica, che vengono diagnosticate e differenziate da specifici modelli biochimici di porfirine e precursori di porfirine nelle urine, nelle feci e nel sangue. Esiste un’ampia gamma di sintomi diversi, anche se la grande maggioranza dei portatori del gene rimane senza sintomi.
Tutte le porfirie sono causate da un disturbo metabolico ereditario della biosintesi dell’ema nel fegato o negli eritrociti. Esiste una modalità di eredità autosomica dominante. La formazione di ema dalla glicina e dal succinil-CoA avviene in 8 fasi enzimatiche, ognuna delle quali può essere influenzata da un difetto genetico. Di conseguenza, si verifica un accumulo di porfirine o dei loro precursori e un aumento dell’escrezione [1,2]. Sebbene solo il 20-30% dei portatori di difetti diventi sintomatico (in genere tra i 20 e i 40 anni), esistono anche forme massive e pericolose per la vita, in particolare nella porfiria acuta intermittente, dove gli attacchi sono accompagnati da una triade di dolore addominale, sintomi cardiologici e neuropsichiatrici [1–3].
Classificazione
Nell’ICD-10, viene fatta una distinzione tra le porfirie eritropoietiche (sito principale di formazione nel midollo osseo) e altre forme di porfiria in base al sito principale di formazione dei prodotti intermedi della sintesi dell’ema [4–6]:
- E80.0: Porfiria eritropoietica ereditaria (incl. porfiria eritropoietica congenita e porfiria eritropoietica)
- E80.1: Porfiria cutanea tarda
- E80.2: Altre porfirie (inclusa la porfiria epatica acuta e la porfiria acuta intermittente)
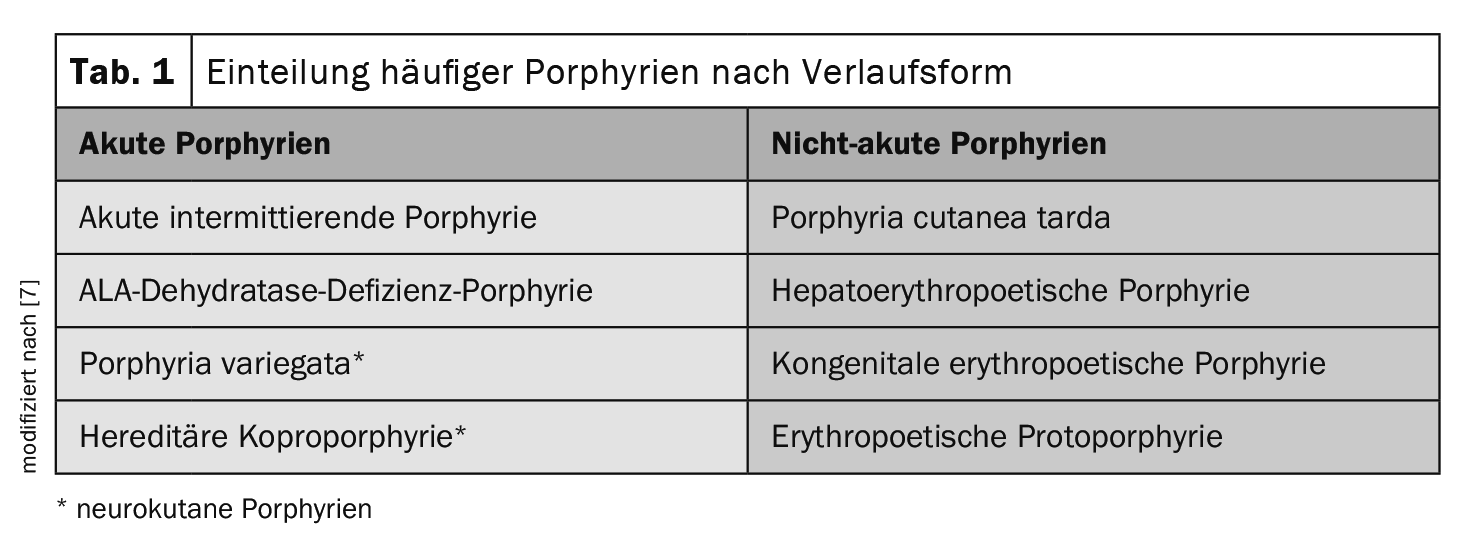
Un altro criterio di categorizzazione è la forma di progressione (Tabella 1) . Questo criterio è più comunemente utilizzato nella pratica medica quotidiana, in quanto tiene conto degli attacchi neuroviscerali acuti potenzialmente pericolosi per la vita [7]:
Nelle porfirie acute, il dolore addominale acuto si presenta a episodi insieme a deficit neurologici (la cosiddetta forma neuro-viscerale). L’urina di colore rossastro è tipica durante gli attacchi acuti. La porfiria acuta più comune è la porfiria acuta intermittente (AIP).
Le porfirie non acute sono caratterizzate dal coinvolgimento della pelle. I prodotti intermedi che si accumulano nella pelle rendono la pelle più sensibile alla luce, con conseguenti cambiamenti tipici come vesciche e cicatrici successive.
Clinica
La porfiria deve essere presa in considerazione in caso di sintomi addominali, ad esempio di natura colica, disturbi della motilità intestinale (vomito, costipazione, anche diarrea) in connessione con adynamia, confusione, cefalea, iponatriemia, alterazione dello stato di coscienza, convulsioni e una polineuropatia motoria accentuata grave e rapidamente progressiva [2]. Quest’ultima è caratterizzata da un decorso grave, rapido e talvolta doloroso, con enfasi motoria e prossimale, talvolta accompagnata da neurite dei nervi cranici e disturbi autonomici. Si ritiene che una serie di farmaci possa scatenare gli attacchi [2].
Diagnosi
La diagnosi di porfiria si basa sul rilevamento di precursori ematici accumulati nelle feci, nelle urine e nel plasma, corrispondenti al difetto enzimatico [1]. Un rilevamento di screening qualitativo del porfobilinogeno è possibile utilizzando il test Hoesch-Schwartz-Watson [2]. Il sospetto clinico di porfiria deve essere confermato mediante test dei metaboliti nelle urine, nelle feci e nel sangue, rilevando i precursori delle porfirine eccessivamente elevati, l’acido δ-aminolevulinico e il porfobilinogeno, nonché le porfirine nelle urine. La diagnosi differenziale delle varie forme di porfiria viene effettuata in una seconda fase su campioni di urina, feci e sangue. A differenza delle porfirie acute, i due precursori delle porfirine non sono elevati nelle porfirie non acute. Le determinazioni enzimatiche e i test genetici molecolari sono possibili per determinare lo stadio del difetto enzimatico, ma non sono rilevanti per la diagnosi clinica e il trattamento.
| Caso clinico In una donna di 28 anni che soffriva di dolori addominali dipendenti dal ciclo, la diagnostica approfondita non ha rivelato alcun risultato conclusivo. È stato quindi consigliato un trattamento psicosomatico. Tuttavia, i sintomi sono peggiorati dopo una cura a base di digiuno: si sono verificate parestesie dolorose alle cosce, nausea, costipazione e vomito. Per alleviare i sintomi è stato prescritto il metamizolo. |
| Nel corso della malattia, il paziente ha sviluppato una paralisi incipiente dei muscoli estensori di entrambe le mani, una grave iponatriemia (concentrazione di sodio 105 mmol/l) e allucinazioni. Questo ha portato al ricovero in ospedale e all’immediata terapia intensiva. Dopo aver trovato urine rossastre nella sacca delle urine e un aumento massiccio delle concentrazioni di ALA e PBG** in un campione di urine, è stata fatta la diagnosi di porfiria epatica acuta (AHP). Le porfirie epatiche acute sono caratterizzate dalla comparsa di attacchi neuro-viscerali con o senza manifestazioni cutanee. L’AHP più comune è la porfiria acuta intermittente. |
| Il paziente è stato poi trattato con emarginato per via endovenosa (3 mg/kgKG in 100 ml di soluzione di albumina al 20% per 30 minuti) per quattro giorni. A causa del vomito, l’apporto calorico doveva essere somministrato per via parenterale tramite infusioni. Nella fase acuta, l’apporto calorico target era di 24 kcal/kg di peso corporeo al giorno. Per quanto riguarda la tolleranza metabolica individuale, sono state effettuate determinazioni giornaliere del fosfato nel siero e controlli della glicemia ogni sei ore, per prevenire la sindrome da refezione. |
| ** ALA=acido delta-aminolevulinico, PBG=porfobilinogeno |
| a [8,9] |
Terapia
In primo luogo, tutti i farmaci porfirinogeni devono essere sospesi e sostituiti con farmaci “compatibili con la porfiria” [2]. Si raccomandano anche le seguenti misure:
Porfirie acute: Negli attacchi gravi e con una diagnosi confermata, si effettua un trattamento con emarginato [6]. Inoltre, è necessario somministrare una quantità sufficiente di glucosio (4-5 g/kg di peso corporeo/d). A seconda delle condizioni del paziente, la somministrazione può avvenire per via orale, tramite sondino nasogastrico o per via endovenosa. La somministrazione orale di glucosio può essere effettuata, ad esempio, con una soluzione di maltodestrina (25%). Anche nei casi asintomatici di porfiria acuta intermittente, è necessario effettuare esami regolari (1-2 volte all’anno) presso un centro epatologico, poiché in questa forma esiste un rischio maggiore di sviluppare un tumore al fegato, come nel caso della porfiria cutanea tarda (PCT). Per la porfiria variegata e la coproporfiria ereditaria, un’adeguata protezione dalla luce e la profilassi delle riacutizzazioni sono tra le misure più importanti [6].
Porfirie non acute: Le porfirie non acute sono caratterizzate da sintomi cutanei [6]. Come regola generale, i pazienti devono evitare l’esposizione diretta della pelle alla luce UV. Nella PCT, l’accumulo eccessivo di ferro può essere ridotto con la flebotomia, che di solito porta a un miglioramento dei sintomi. Un altro approccio terapeutico è la somministrazione del farmaco antimalarico clorochina [6]. Nel trattamento della protoporfiria eritropoietica, una riduzione efficace della fotosensibilità può essere ottenuta con l’assunzione quotidiana di betacarotene da febbraio a ottobre.
Letteratura:
- Strato P, Rosien U (eds.). Fegato. Gastroenterologia pratica. 2011:281-366.
- Straube A, et al: Disturbi metabolici. NeuroIntensiv 2015 Apr 30: 643-723.
- Kauppinen R: Porfirie. Lancet 2005; 365: 241-252.
- Classificazione Statistica Internazionale delle Malattie e dei Problemi Sanitari Correlati: ICD-10, www.icd-code.de, (ultimo accesso 09.11.2023)
- Flexikon, https://flexikon.doccheck.com/de/Porphyrie,(ultimo accesso 09.11.2023)
- Ospedale universitario di Düsseldorf, www.uniklinik-
duesseldorf.de/patienten-besucher/klinikeninstitutezentren/klinik-fuer-gastroenterologie-hepatologie-und-infektiologie/klinik/fuer-patienten/behandlungsschwerpunkte/metabolic-diseases/porphyria, (ultimo accesso 09.11.2023) - Muschalek W, et al: Le porfirie. JDDG 2022; (20, Numero 3): 316-333.
- Stölzel U, Stauch T, Kubisch I. Porfirie [Porphyria]. Internist (Berl) 2021; 62(9): 937-951.
- orphanet: Porfiria epatica acuta, www.orpha.net,(ultimo accesso 09.11.2023).
PRATICA GP 2023: 18(11): 48-49









