Le vieillissement de la population entraîne une augmentation des cas de SMD. Les développements dans le domaine du diagnostic moléculaire et les nouvelles thérapies vont toutefois également modifier la prise en charge des patients âgés. Il est important d’avoir une approche et des échanges multidisciplinaires.
Les syndromes myélodysplasiques (SMD) sont diagnostiqués en tant que maladies du sujet âgé, principalement chez les patients >70 ans. En Suisse, avec une incidence de 2-3/100’000 patients-années, on peut s’attendre à un peu plus de 300 nouveaux cas par an. On estime qu’il y a actuellement environ 1600 patients atteints de SMD dans notre pays [1]. Les options thérapeutiques sont restées essentiellement les mêmes ces dernières années, avec la transplantation de cellules souches allogéniques comme traitement curatif, possible pour quelques patients seulement, et différentes options pour améliorer les cytopénies dans les situations palliatives restantes [2–5]. En revanche, le développement rapide du séquençage de “nouvelle génération” (NGS) a également conduit à de nouvelles découvertes pertinentes dans le quotidien clinique de l’hématologie [6,7], qui jouent un rôle pour les patients SMD en termes de diagnostic et d’évaluation du pronostic. Sur la base de la classification de l’OMS mise à jour en 2016, les principaux développements et concepts sont présentés ci-dessous.
Révision de la classification de l’OMS 2016
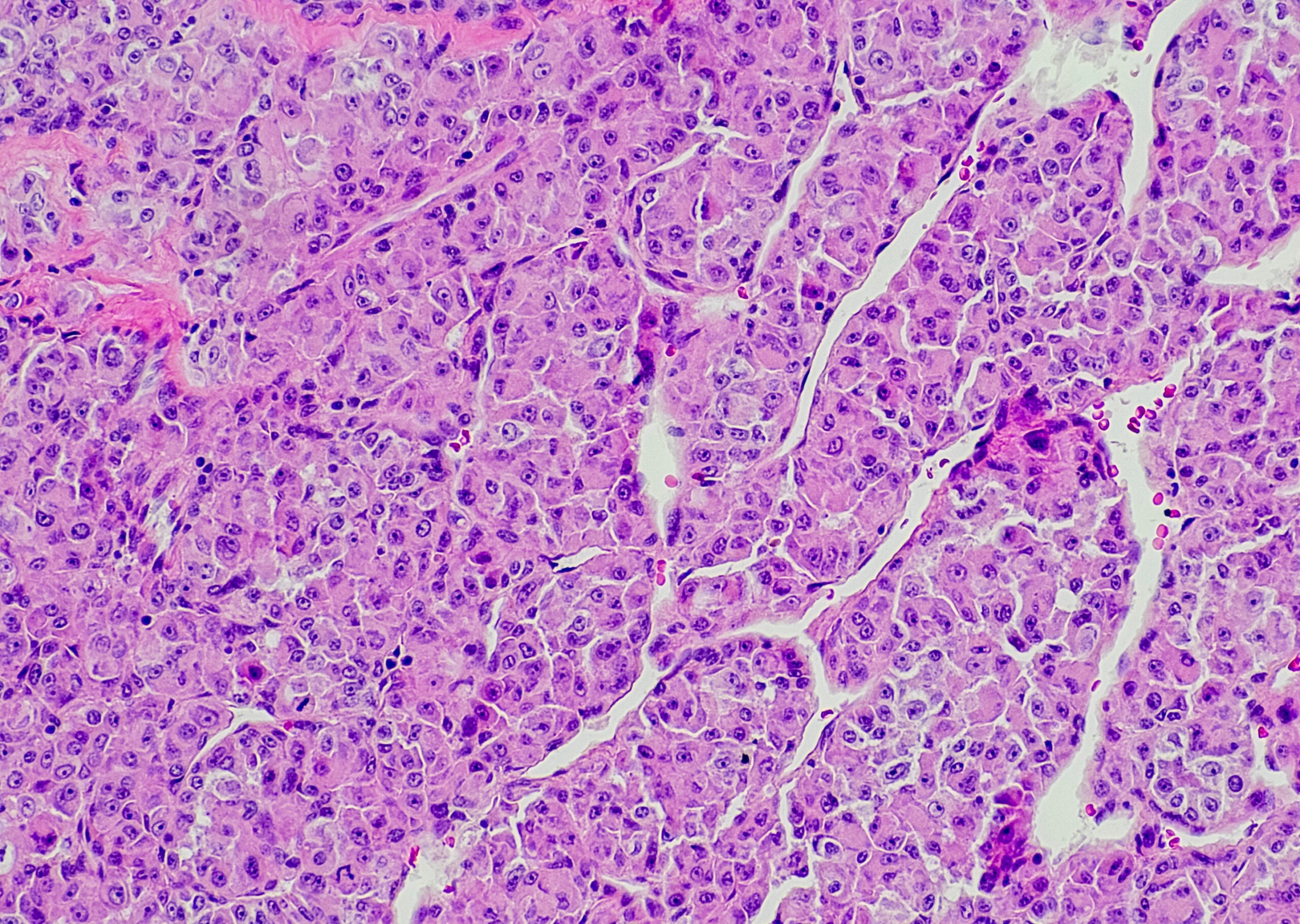
Même à l’ère de la biologie moléculaire, l’évaluation morphologique des frottis de sang périphérique et des aspirations de moelle osseuse reste la base du diagnostic. On distingue fondamentalement les formes de SMD avec excès de blastes de celles sans prolifération de blastes. Le nombre de rangées de cellules affectées par les cytopénies et les dysplasies, la détection de sidéroblastes en anneau (RS) et les modifications cytogénétiques typiques sont déterminants pour la subdivision ultérieure. L’une des nouveautés de la classification OMS 2016 réside dans la nomenclature (tab. 1) [8,9]. Les termes “anémie réfractaire” et “cytopénie réfractaire” ont été abandonnés et le terme “syndrome myélodysplasique” est désormais utilisé pour toutes les entités, complété par le résultat morphologique principal. Cela corrige certaines incohérences de l’ancienne terminologie, comme le terme “anémie réfractaire” pour les formes de SMD avec excès de blastes souvent associées à une pancytopénie.

La cytogénétique conventionnelle des métaphases reste le deuxième pilier indispensable du diagnostic des SMD. Chez les patients présentant des cytopénies sans prolifération de blastes et sans dysplasie (significative), le diagnostic peut être établi par la mise en évidence de certaines anomalies cytogénétiques définissant les SMD (tableau 2). Dans ce cas, le diagnostic de SMD inclassable est attribué. Sont également classés dans cette catégorie les cas de pancytopénie et de dysplasie unilinéaire ou de détection constante de blastes à 1% dans le sang périphérique sans multiplication de blastes dans la moelle osseuse [8,9].
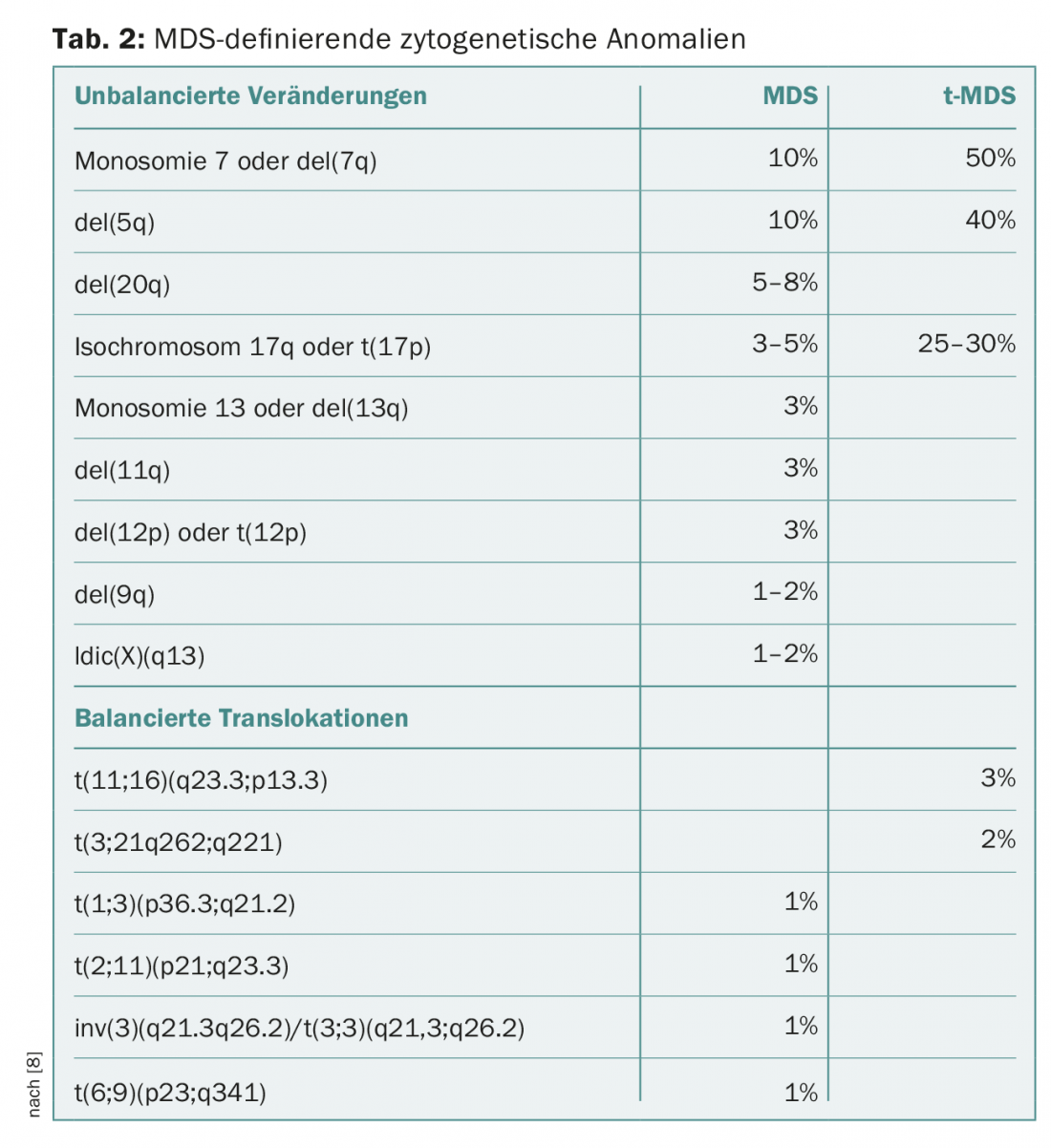
La catégorie des SMD avec del(5q), caractérisée par une délétion sur le bras court du chromosome 5, a été élargie dans sa définition. Désormais, les cas présentant une deuxième anomalie cytogénétique peuvent également être classés dans cette catégorie, sauf en cas d’anomalies supplémentaires du chromosome 7, qui sont associées à un pronostic nettement moins bon [8–10].
La cytogénétique conventionnelle (l’analyse d’au moins 20 métaphases est requise pour un examen congruent) peut être complétée par d’autres méthodes dans des situations sélectionnées, par exemple en cas de forte suspicion morphologique de SMD del(5q), mais de caryotype normal. Les panels FISH focalisés sur les anomalies chromosomiques typiques des SMD ou les microarrays d’hybridation génomique comparative (array-CGH) à l’échelle du génome peuvent être utilisés à cet effet [11]. Ces deux méthodes ne constituent toutefois pas un remplacement a priori de la cytogénétique conventionnelle. En particulier, la valeur pronostique des anomalies détectables uniquement dans l’array CGH n’est pas encore établie avec certitude à l’heure actuelle.
Importance diagnostique du séquençage “nouvelle génération” dans les cytopénies indéterminées
Ces dernières années, de nombreuses mutations ont été identifiées dans des gènes associés à l’apparition des SMD et à la progression vers la LAM. Le spectre des “gènes pilotes” mutés est varié et comprend souvent des composants de la modification épigénétique de l’ADN et des histones ou de la machinerie d’épissage modifiant l’ARN (“spliceosome”). Des composants de la régulation du cycle cellulaire, des complexes de cohésine, des facteurs de transcription ou des composants de la transduction du signal intracellulaire peuvent également être affectés (tableau 3) [2,12–18].
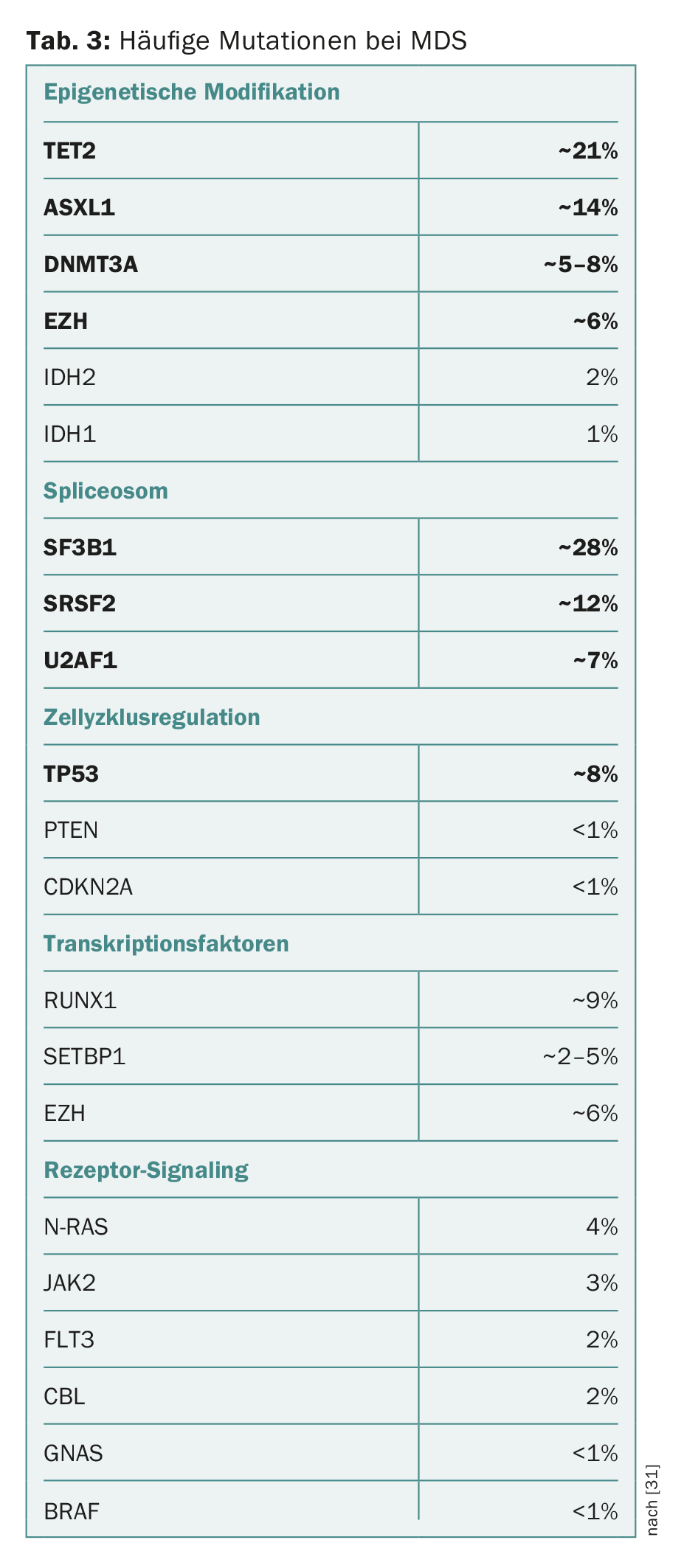
Il convient de souligner, d’une part, que la plupart des mutations du gène driver observées dans le SMD se retrouvent également dans d’autres néoplasies myéloïdes, bien que dans des fréquences ou des combinaisons différentes. En outre, plusieurs études ont montré que des mutations typiques des néoplasies myéloïdes peuvent également se produire chez des personnes hématologiquement saines. <La fréquence augmente fortement avec l’âge, de 10% chez les personnes de 60 ans à 15-20% chez les personnes de >80 ans (mais <1% chez les personnes de 40 ans). Ce phénomène a été appelé “Clonal Hematopoiesis of Indeterminate Potential” (CHIP) [19,20]. Comme la gammapathie monoclonale de signification indéterminée (MGUS) et la lymphocytose monoclonale à cellules B (MBL), il s’agit d’une précancérose facultative qui peut évoluer vers une maladie hématologique maligne à raison d’environ 1% par an. De plus, les patients atteints de CHIP présentent également une morbidité cardiovasculaire accrue [21].
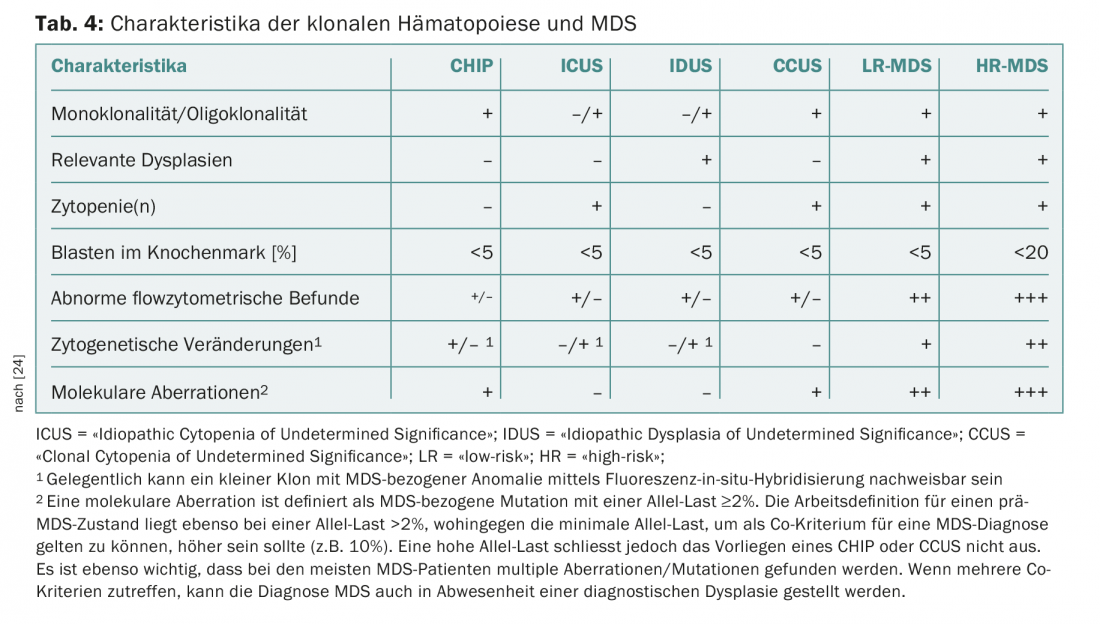
Un problème récurrent en pratique et en clinique est celui des patients présentant des cytopénies persistantes pour lesquelles aucune cause certaine ne peut être trouvée. En l’absence de dysplasies ou d’anomalies cytogénétiques définissant le SMD, celles-ci sont jusqu’à présent regroupées sous le terme de “cytopénie idiopathique de signification indéterminée” (ICUS) [22]. La détection de mutations récurrentes peut ici aider à distinguer les cytopénies réactives des cytopénies clonales. L’interprétation d’une détection de mutation dépend alors de la taille du clone (mesurée par la “variant allele frequency”, VAF) et du nombre de mutations détectées. Si une mutation récurrente avec une FAV supérieure à 2% est trouvée dans une constellation ICUS, on parle de “cytopénie clonale de signification indéterminée” (CCUS). Parmi les patients atteints de CCUS, ceux qui présentent au moins deux mutations avec une FAV >10% ont un risque élevé de développer une néoplasie hématologique dans les cinq prochaines années [23]. Les critères récemment présentés par un panel international d’experts pour distinguer les CHIP, ICUS, CCUS d’un MDS manifeste [24] sont résumés dans le tableau 4. Ce consensus définit également de nouveaux critères mineurs liés aux SMD qui peuvent être utilisés dans des situations non concluantes pour établir un diagnostic provisoire de SMD (tab. 5).

L’accumulation séquentielle de modifications génétiques dans les CSH a longtemps été postulée comme corrélat pathogénique de l’évolution clonale et le concept d’hématopoïèse clonale démontre un chevauchement entre les modifications de l’hématopoïèse au cours du vieillissement et la pathogenèse des néoplasies myéloïdes. Cependant, les facteurs responsables de la transition de la CHIP et de la CCUS vers une néoplasie manifeste ne sont pas clairs. L’évolution clonale semble être conditionnée non seulement par des mécanismes intrinsèques à la cellule (mutations dans les cellules souches hématopoïétiques), mais aussi par des mécanismes extrinsèques à la cellule. Le microenvironnement de la moelle osseuse et les composants du système immunitaire inné et acquis jouent un rôle à cet égard. Le “stress immunologique” explique probablement aussi l’association avec des maladies ou des phénomènes inflammatoires et immunologiques concomitants, en partie inclassables, qui peuvent survenir chez les patients atteints de SMD [25–28]. La perte de contrôle immunologique de la tumeur ainsi que les modifications favorisantes dans la niche de la moelle osseuse sont actuellement au centre de la recherche fondamentale. Cela conduira peut-être à l’avenir à de nouvelles approches thérapeutiques qui pourraient être utilisées très tôt dans le développement des néoplasies myéloïdes (Fig. 1).
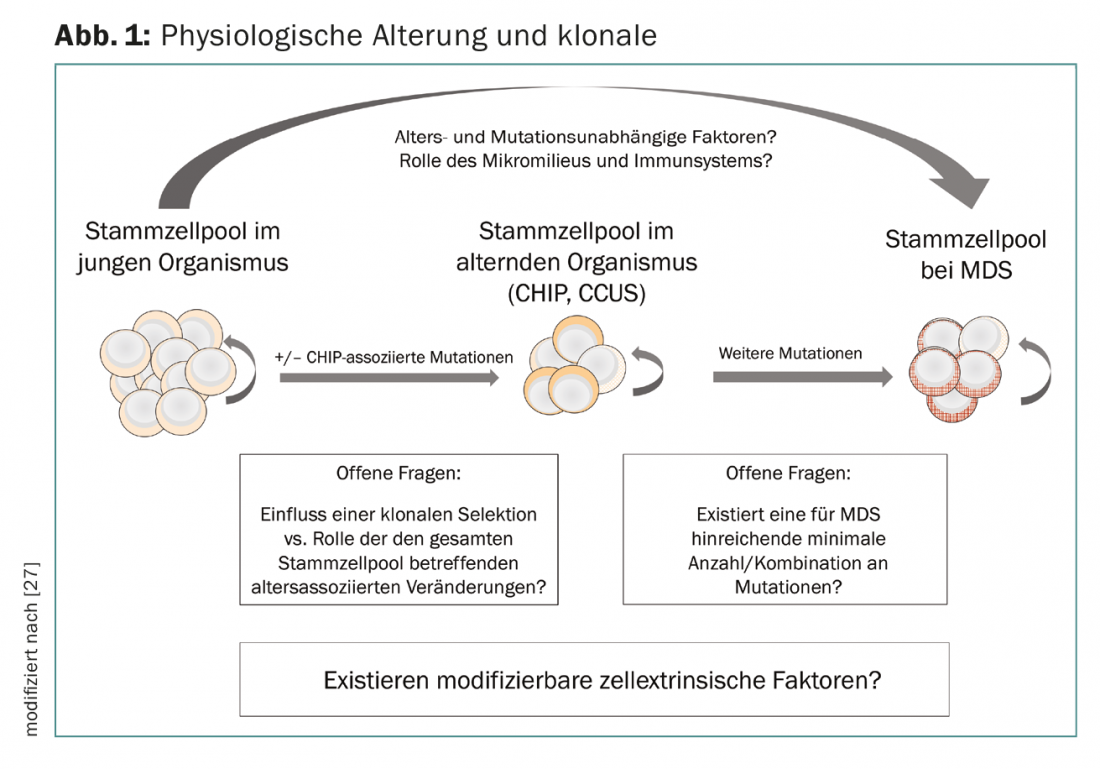
En raison du chevauchement dans le spectre des mutations entre CHIP, CCUS et les SMD, les analyses des mutations n’ont délibérément pas été reprises dans la classification actuelle de l’OMS. Les mutations dans le gène driver de la composante SF3B1 du spliceosome, qui sont très fortement associées à un phénotype ringsideroblastique, constituent une exception [29]. En cas de mutation SF3B1, selon l’OMS 2016, la détection de 5% de RS suffit pour être classé dans le groupe des SMD avec RS, au lieu des 15% normalement requis. Les patients mutés SF3B1 ont un très bon pronostic avec une faible probabilité de progression vers une LAM. Comme les patients atteints de dysplasie multiligne et de RS bénéficient également de l’influence pronostique d’une mutation SF3B1, l’entité SMD avec dysplasie multiligne et RS a été réintroduite dans la classification de 2016.
Importance pronostique et prédictive supplémentaire des mutations du gène driver
D’un point de vue pronostique, on peut d’ores et déjà identifier quelques autres scénarios cliniques dans lesquels l’analyse des mutations peut fournir des informations pertinentes. Environ 15% des patients atteints de SMD del(5q) présentent une mutation TP53. Bien que ces derniers répondent également au lénalidomide sur le plan hématologique, ils obtiennent plus rarement une rémission cytogénétique et présentent un risque plus élevé de transition vers une LAM. En cas de détection d’une mutation de TP53 dans le SMD del(5q), des alternatives thérapeutiques peuvent donc être envisagées [3,30].
Un autre scénario de recherche de mutations concerne le groupe hétérogène de patients du groupe “intermédiaire” selon l’IPSS-R. Il s’agit d’un groupe de patients dont la maladie est diagnostiquée à un stade précoce. Ceux-ci peuvent être traités soit selon les recommandations applicables aux patients “à faible risque” ou “à haut risque”, en fonction de la présence ou de l’absence d’autres caractéristiques de risque [2–5,30]. Jusqu’à présent, outre l’examen isolé de la cytogénétique (constellation à haut risque ?), on ne dispose à cet effet que d’autres marqueurs de risque conventionnels (p. ex. élévation de la LDH, fibrose de la moelle osseuse >degré 2 selon l’OMS). Plusieurs mutations récurrentes sont associées à un risque significativement plus élevé et justifient un upstaging pronostique dans la catégorie “high-risk” et, chez des patients sélectionnés, une allogreffe de cellules souches [15]. La recherche d’une mutation TP53, ASXL1, RUNX1, EZH2 et ETV6 chez les patients éligibles à un traitement intensif est donc explicitement recommandée dans les guidelines actuelles [4,15]. Un projet international parrainé par la MDS Foundation s’efforce actuellement de développer un “IPSS-R moléculaire” complété par le statut mutationnel.
Les données cliniquement utilisables sur la valeur prédictive du profil mutationnel dans le SMD sont actuellement limitées. Une pertinence pour la stratification des thérapies ciblées se dessine pour l’avenir. Le luspatercept (ACE-356) est un inhibiteur du TGF-β-superfamilial [10], qui a montré un taux de réponse érythroïde élevé chez les patients SMD réfractaires à l’EPO présentant une RS et/ou des mutations dans SF3B1. En outre, des inhibiteurs de spliceosome (H3B-8800) sont actuellement à l’étude dans la LAM et les SMD, qui éliminent préférentiellement les clones présentant des mutations dans les gènes conducteurs de spliceosome en raison de l’insuffisance haplo (effet cyclope). En outre, la midostaurine (mutations FLT3), l’enasidenib (mutations IDH2) et l’ivosidenib (mutations IDH1), déjà approuvés dans la LAM, sont actuellement en développement clinique dans les SMD à haut risque présentant un profil de mutations correspondant, que ce soit en tant que substance unique ou en association avec un traitement hypométhylant (HMT) ou une chimiothérapie standard.
Messages Take-Home
- En raison du vieillissement de notre société, on peut s’attendre à une augmentation significative des SMD.
- Les développements dans le domaine du diagnostic moléculaire ainsi que les nouvelles options thérapeutiques vont modifier les stratégies de traitement, y compris pour les patients âgés.
- La gestion multidisciplinaire est une condition essentielle et pose de nouveaux défis aux systèmes de santé. Cela ne concerne pas seulement les soins adéquats dans la routine clinique, mais aussi la réalisation d’études cliniques qui, dans le cas des maladies rares, exigent un haut degré de coopération et de coordination à l’ère de la médecine personnalisée.
- En réponse à ces défis, le groupe d’étude suisse des SMD a créé en 2015 le registre/biobanque suisse des SMD afin de permettre les échanges cliniques et scientifiques au sein d’un réseau international.
Littérature :
- Bonadies N, et al : Trends of classification, incidence, mortality, and survival of MDS patients in Switzerland between 2001 and 2012. Cancer epidemiology 2017 ; 46 : 85-92.
- Malcovati L, et al : Diagnostic et traitement des syndromes myélodysplasiques primaires chez l’adulte : Recommandations du réseau européen LeukemiaNet. Blood 2013 ; 122(17) : 2943-2964.
- Fenaux P, et al : Myelodysplastic syndromes : ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology : official journal of the European Society for Medical Oncology 2014 ; 25(Suppl 3) : iii57-69.
- Hofmann WK, Platzbecker U, Götze K : Ligne directrice d’Onkopedia sur les syndromes myélodysplasiques. Situation en mars 2016.
- Greenberg PL, et al : Syndromes myélodysplasiques, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network : JNCCN 2017 ; 15(1) : 60-87.
- Johnsen JM, Nickerson DA, Reiner AP : Massively parallel sequencing : The new frontier of hematologic genomics. Blood 2013 ; 122(19) : 3268-3275.
- Kuo FC, et al : The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood 2017 ; 130(4) : 433-439.
- Swerdlow SH, et al. (éd.) : WHO classification of tumours of haematopoietic and lymphoid tissues. 4e édition révisée. Lyon : Centre international de recherche sur le cancer 2017.
- Arber DA, et al : La révision 2016 de la classification de l’Organisation mondiale de la santé des néoplasmes myéloïdes et des leucémies aiguës. Blood 2016 ; 127(20) : 2391-2405.
- Mies A, Platzbecker U : Increasing the effectiveness of hematopoiesis in myelodysplastic syndromes : Erythropoiesis-stimulating agents and transforming growth factor-β superfamily inhibitors. Seminars in hematology 2017 ; 54(3) : 141-146.
- Ouahchi I, et al : Microarray-based comparative genomic hybridation reveals additional recurrent aberrations in adult patients evaluated for myelodysplastic syndrome with normal karyotype. British journal of haematology 2018. DOI : 10.1111/bjh.15068 [Epub ahead of print].
- Kon A, et al : Mutations récurrentes dans de multiples composants du complexe de la cohesine dans les néoplasmes myéloïdes. Nature genetics 2013 ; 45(10) : 1232-1237.
- Yoshida K, et al : Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011 ; 478(7367) : 64-69.
- Bejar R, Levine R, Ebert BL : Unraveling the molecular pathophysiology of myelodysplastic syndromes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011 ; 29(5) : 504-515.
- Bejar R, et al : Effet clinique des mutations de points dans les syndromes myélodysplasiques. The New England journal of medicine 2011 ; 364(26) : 2496-2506.
- Abdel-Wahab O, Figueroa ME : Interpreting new molecular genetics in myelodysplastic syndromes. Hematology American Society of Hematology Education Program 2012 ; 2012 : 56-64.
- Leeke B, et al : Mutations de la cohésine dans les malignités myéloïdes : mécanismes sous-jacents. Experimental hematology & oncology 2014 ; 3 : 13.
- Tothova Z, Steensma DP, Ebert BL : Nouvelles stratégies dans les syndromes myélodysplasiques : Application du diagnostic moléculaire à la pratique clinique. Clinical cancer research : an official journal of the American Association for Cancer Research 2013 ; 19(7) : 1637-1643.
- Jan M, Ebert BL, Jaiswal S : Hématopoïèse clonale. Seminars in hematology 2017 ; 54(1) : 43-50.
- Heuser M, Thol F, Ganser A : Hématopoïèse clonale de potentiel indéterminé. Deutsches Arzteblatt international 2016 ; 113(18) : 317-322.
- Fuster JJ, Walsh K : Mutations somatiques et hématopoïèse clonale : nouveaux facteurs potentiels inattendus de maladies cardiovasculaires liées à l’âge. Circulation research 2018 ; 122(3) : 523-532.
- Valent P, et al : Cytopénie idiopathique de signification indéterminée (CIUS) et dysplasie idiopathique de signification incertaine (IDUS), et leur distinction des SMD à faible risque. Leukemia research 2012 ; 36(1) : 1-5.
- Malcovati L, et al : Signification clinique de la mutation somatique dans la cytopénie sanguine inexpliquée. Blood 2017 ; 129(25) : 3371-3378.
- Valent P, et al : Proposed minimal diagnostic criteria for myelodysplastic syndromes (MDS) and potential pre-MDS conditions. Oncotarget 2017 ; 8(43) : 73483-73500.
- Gañán-Gómez I, et al. : Dérégulation de la signalisation immunitaire et inflammatoire innée dans les syndromes myélodysplasiques. Leucémie 2015 ; 29(7) : 1458-1469.
- Glenthøj A, et al : Mécanismes immunitaires dans le syndrome myélodysplasique. International journal of molecular sciences 2016 ; 17(6) : 944.
- Chung SS, Park CY : Vieillissement, hématopoïèse et syndromes myélodysplasiques. Blood advances 2017 ; 1(26) : 2572-2578.
- Cooper JN, Young NS : Clonality in context : Hematopoietic clones in their marrow environment. Blood 2017 ; 130(22) : 2363-2372.
- Malcovati L, et al : La mutation SF3B1 identifie un sous-ensemble distinct du syndrome myélodysplasique avec sidéroblastes annulaires. Blood 2015 ; 126(2) : 233-241.
- Nordic MDS Study Group : Guidelines to Patient Management of Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia. 8e mise à jour. www.nmds.org/index.php/guidelines (cité le 14.03.2018).
- Montalban-Bravo G, Garcia-Manero G : Myelodysplastic syndromes : 2018 update on diagnosis, risk-stratification and management. Am J Hematol 2018 Jan ; 93(1) : 129-147.
Littérature complémentaire :
- List A, Ebert BL, Fenaux P : Une décennie de progrès dans le syndrome myélodysplasique avec délétion du chromosome 5q. Leucémie 2018. DOI : 10.1038/s41375-018-0029-9 [Epub ahead of print].
- Platzbecker U, et al : Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS) : A multicentre, open-label phase 2 dose-finding study with long-term extension study. The Lancet Oncology 2017 ; 18(10) : 1338-1347.
InFo ONKOLOGIE & HÉMATOLOGIE 2018 ; 6(2) : 22-26