Les dermatoses bulleuses auto-immunes constituent un groupe hétérogène de maladies auto-immunes rares, parfois graves, parmi lesquelles on trouve le pemphigus et la pemphigoïde, l’épidermolyse bulleuse acquise et la dermatite herpétiforme de Duhring. Les caractéristiques communes des dermatoses bulleuses auto-immunes – à l’exception de la maladie de Duhring – sont des auto-anticorps dirigés contre les protéines structurelles de la peau et des muqueuses et responsables d’une perte d’intégrité cutanée [1].
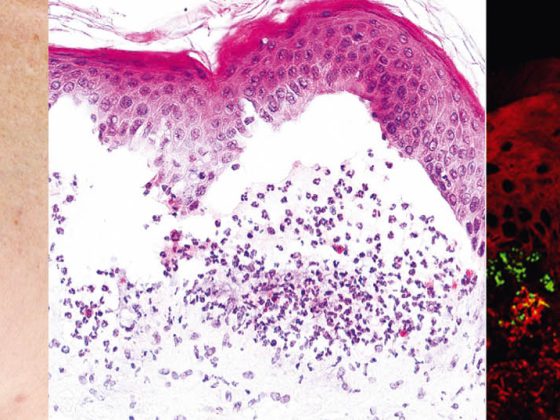
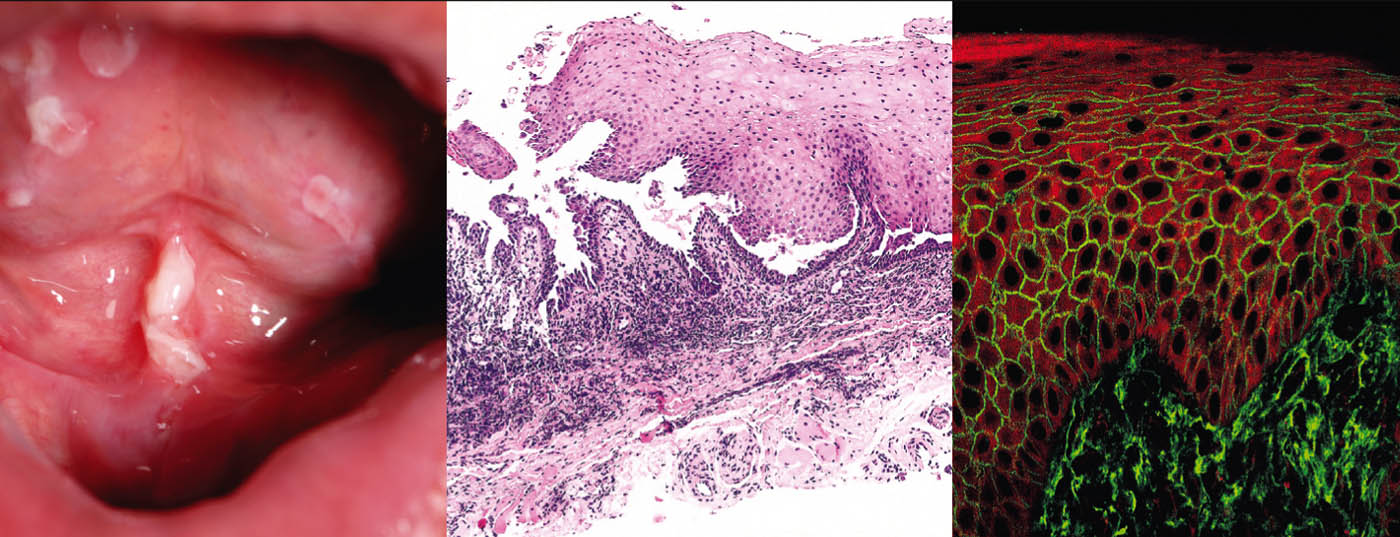
Le pemphigus vulgaire (tab. 1, fig. 1) se manifeste par des bulles mucocutanées généralisées et flasques qui se rompent très rapidement, de sorte que le tableau clinique est généralement dominé par des érosions et des croûtes.

La plupart des patients présentent une atteinte des muqueuses, en particulier dans la bouche, qui est très douloureuse, et il n’est donc pas rare que le diagnostic initial soit posé par les dentistes. Du point de vue pathogénique, le pemphigus vulgaire est caractérisé par des auto-anticorps (AK) dirigés contre les protéines d’adhésion cellulaire – les desmogléines 1 et 3. Chez les patients atteints exclusivement par voie orale, les AK sont dirigées contre la desmogléine 3. L’histopathologie révèle un clivage intraépidermique suprabasal avec acantholyse et formation de “pierres tombales” des kératinocytes basaux. L’immunofluorescence directe (DIF) montre des dépôts intercellulaires typiques d’IgG et de C3 dans l’épiderme. L’immunofluorescence indirecte (IIF) à partir du sérum du patient confirme la présence d’autoanticorps IgG circulants. Le test ELISA permet de détecter directement la desmogléine 3 et la desmogléine 1 dans le sérum du patient [2].
Le traitement vise principalement à réduire la production d’auto-anticorps. Dans ce cas, les corticostéroïdes systémiques et d’autres immunosuppresseurs tels que l’azathioprine, le mycophénolate mofétil, le cyclophosphamide ou la ciclosporine restent au premier plan des traitements standard. Les autres options sont la plasmaphérèse ou les immunoglobulines intraveineuses (IgIV). Dans les cas résistants au traitement, le rituximab, un anticorps anti-CD20, est notamment une alternative prometteuse. Outre le pemphigus vulgaire, le pemphigus foliacé [3], le pemphigus herpétiforme [4], le pemphigus paranéoplasique [5] et le pemphigus à IgA font également partie du groupe des maladies pemphigus.
Pemphigoïde bulleuse
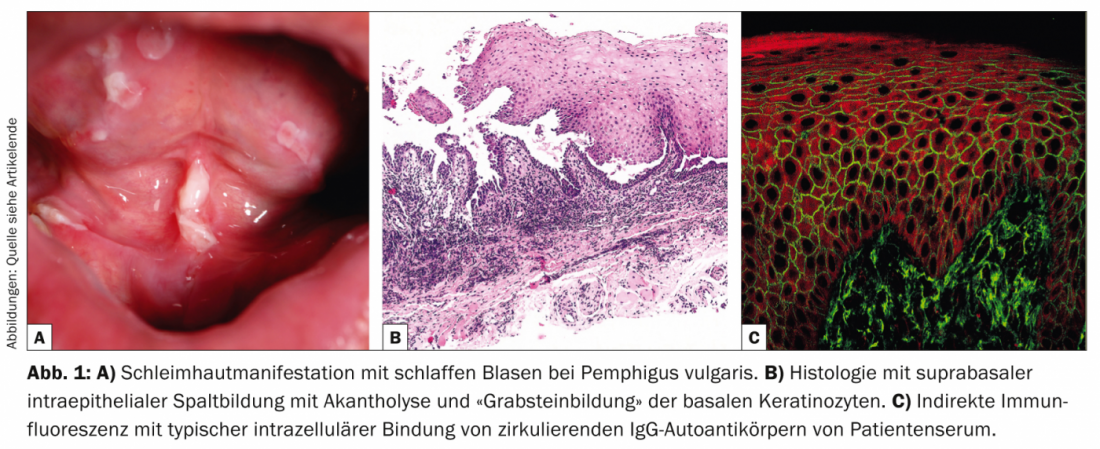
Avec une incidence de 12,1 nouveaux cas/million/an en Suisse, la pemphigoïde bulleuse (tab. 2, fig. 2) est la maladie la plus fréquente du groupe des pemphigoïdes et en même temps la dermatose auto-immune vésicante la plus fréquente [6].

La pemphigoïde bulleuse survient généralement à un âge avancé et se caractérise par des cloques rebondies sur une peau inflammatoire et rouge ou normale qui démange intensément. Souvent, cette affection évolue initialement sans formation de cloques et est diagnostiquée comme eczéma, urticaire ou prurigo en raison du prurit prononcé. D’un point de vue pathogénique, la maladie est due à des auto-anticorps dirigés contre la BP 180 (également connue sous le nom de collagène de type XVII). L’histologie révèle une formation de bulles sous-épidermiques avec un épiderme intact et des infiltrats proéminents riches en éosinophiles. Dans le DIF de la peau périlésionnelle, on observe des dépôts d’IgG le long de la zone de jonction dermo-épidermique (membrane basale), ce qui peut être confirmé dans l’IIF par la détection d’anticorps IgG se liant à la membrane basale. Le taux sérique d’auto-anticorps anti-BP180 est en corrélation avec l’activité de la maladie et peut être déterminé au cours de l’évolution afin de déterminer les besoins ultérieurs en matière de traitement.
Les corticostéroïdes topiques ou systémiques restent la base du traitement. Il existe également des tétracyclines avec des nicotinamides, de la dapsone ou, en cas de maladie grave, des immunosuppresseurs, en particulier l’azathioprine, ainsi que des IgIV et du rituximab dans les cas résistants au traitement.
Dermatose bulleuse à IgA linéaire
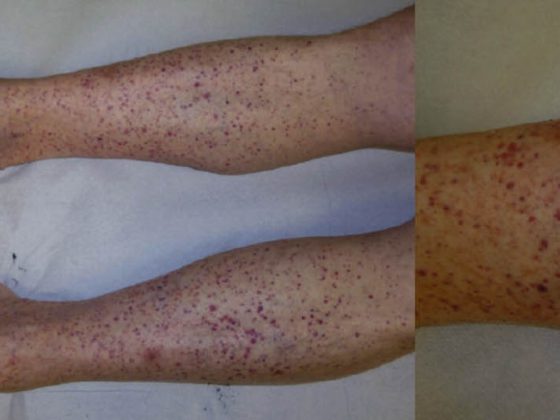
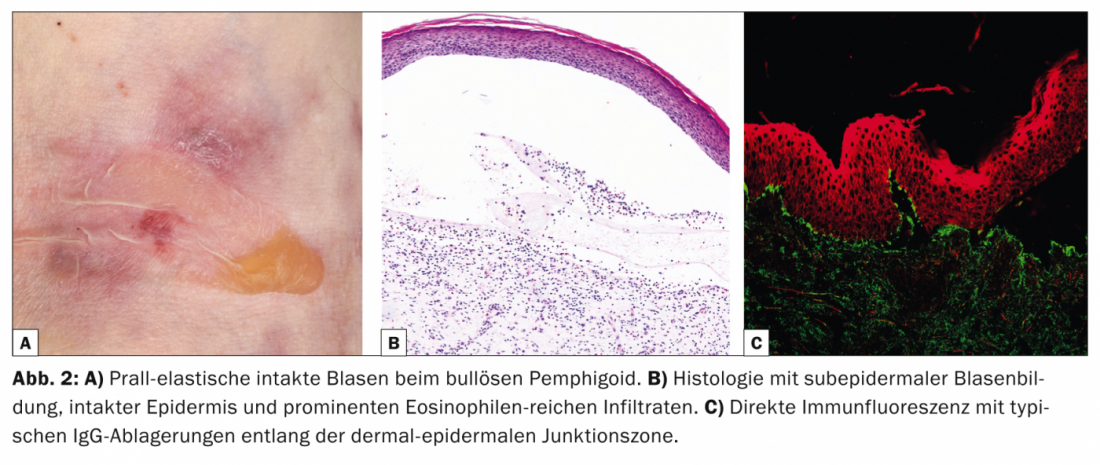
La dermatose linéaire à IgA (LABD, tab. 3, fig. 3) se caractérise par des vésicules et des bulles prurigineuses, généralisées et accentuées sur le côté droit. La maladie touche aussi bien les adultes que les enfants.

Chez l’enfant, elle représente la dermatose bulleuse auto-immune la plus fréquente et évolue généralement de manière autolimitée [7]. Chez l’adulte, la LABD est souvent associée à des médicaments (vancomycine) [8]. Histologiquement, la LABD se caractérise, comme la maladie de Duhring, par une formation de bulles sous-épidermiques, un épiderme intact et des infiltrats riches en neutrophiles au niveau de la zone de jonction dermo-épidermique. Dans le DIF, des dépôts linéaires d’IgA apparaissent dans la zone de la membrane basale.
L’IIF peut confirmer le diagnostic par la détection d’auto-anticorps IgA circulants qui se lient au toit de la vessie. La majorité des patients répondent à un traitement à la dapsone ou aux sulfapyridines. Des traitements réussis ont par ailleurs été décrits chez les enfants et les adultes avec différents antibiotiques (cicloxacilline, érythromycine, tétracycline ou triméthoprime-sulfaméthoxazole).
Epidermiolyse bulleuse aquisita
L’épidermiolyse bulleuse (EBA, tab. 4, fig. 4) se divise cliniquement en une forme localisée, souvent à prédominance acrale, non inflammatoire, cicatricielle et mécano-bulleuse, et en une variante généralisée, inflammatoire et non cicatricielle. La maladie survient généralement à l’âge adulte moyen et est très rare chez les enfants.

Il existe une association étroite avec les maladies inflammatoires chroniques de l’intestin [9]. Sur le plan pathogénique, l’AFP se caractérise par le dépôt d’auto-anticorps IgG contre le procollagène de type VII de la fibrille d’ancrage. Histologiquement, la préparation de routine montre une bulle sous-épidermique avec un épiderme intact. Dans le DIF de la peau périlésionnelle, on trouve des anticorps IgG en bande le long de la zone de jonction dermo-épidermique. Dans l’IIF sur peau de fente humaine séparée par NaCl, qui est positive dans 50% des cas, les auto-anticorps circulants IgG ou plus rarement IgA se lient au fond de la vessie.
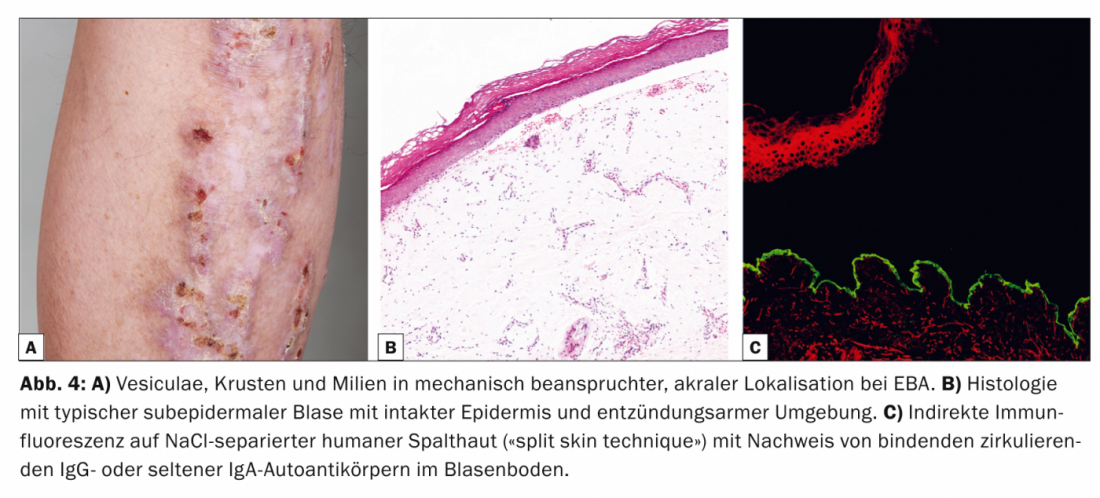
Le Westernblot et l’ELISA peuvent être utilisés en complément pour détecter les autoanticorps IgG circulants [10]. Le traitement de l’AFP est difficile, souvent insatisfaisant et vise principalement à obtenir un effet immunosuppresseur. Les options de traitement actuelles sont donc en premier lieu les corticostéroïdes systémiques, la dapsone ou la colchicine. En outre, dans les cas résistants au traitement, des rapports font état d’une réponse positive au rituximab, un anticorps anti-CD20.
Dermatite herpétiforme
La dermatite herpétiforme (DH, tab. 5, fig. 5) est une manifestation cutanée rare de l’entéropathie sensible au gluten (maladie cœliaque) et se présente sous la forme de papules érythémateuses et de papulovésicules qui démangent violemment et qui apparaissent surtout sur le côté de l’extension et sur le sacrum.

D’un point de vue pathogénique, la DH et la maladie cœliaque sont toutes deux associées au génotype HLA-DQ2, dans lequel des auto-anticorps IgA dirigés contre la réticuline, l’endomysium et la transglutaminase tissulaire ou l’endomysium sont présents. transglutaminase épidermique. Histologiquement, on observe un clivage sous-épidermique et des infiltrats riches en neutrophiles avec parfois la formation de microabcès [11].
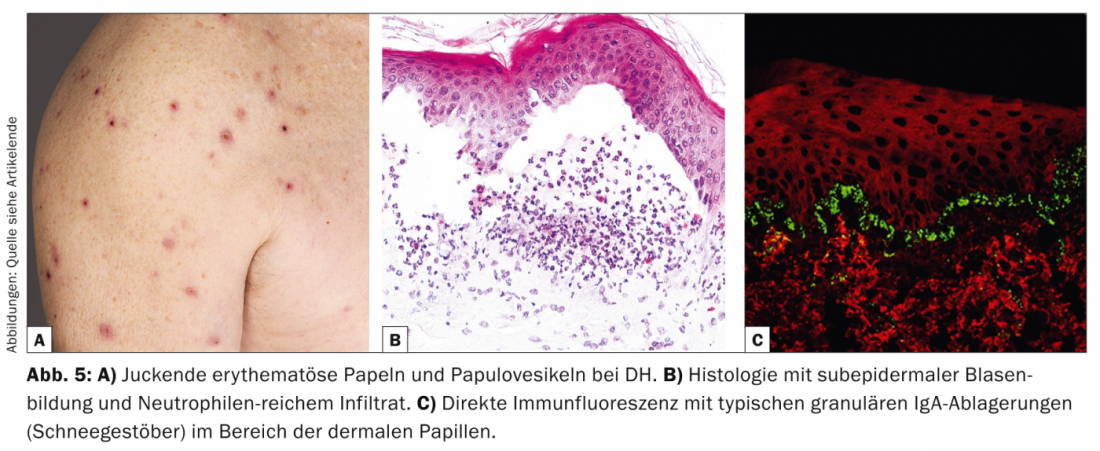
Histologiquement, la DH ressemble à la dermatose bulleuse à IgA linéaire, mais dans la DIF de la peau périlésionnelle, on trouve des dépôts granulaires d’IgA (neige) dans la zone des papilles dermiques. Sur le plan thérapeutique, la priorité est donnée au régime sans gluten, qui améliore à la fois les troubles gastro-entérologiques et la symptomatologie cutanée [12]. La dapsone, les sulfapyridines ou les corticostéroïdes systémiques constituent des options alternatives. Si la dapsone entraîne une amélioration des lésions cutanées, elle n’améliore pas les troubles intestinaux.
Dr. med. Emmanuella Guenova
Sources des images :
Images cliniques : Archives photographiques de l’Hôpital universitaire de Zurich
Images histologiques : Dr. med. Emmanuella Guenova
Images d’immunofluorescence : Birgit Fehrenbacher
Littérature :
- Bolognia, JL, Jorizzo JL, Schaffer JV : Dermatologie. 2012 : Elsevier Health Sciences UK.
- Chan LS : Blistering Skin Diseases. 2009 : Taylor & Francis.
- Guenova E, et al. : Tinea incognito hidden under apparent treatment-resistant pemphigus foliaceus. Acta Derm Venereol 2008 ; 88(3) : 276-277.
- Lebeau S, et al. : Pemphigus herpétiforme : analyse du profil des auto-anticorps au cours de la maladie avec des changements dans le phénotype clinique. Clin Exp Dermatol 2010 ; 35(4) : 366-372.
- Heizmann M, et al : Successful treatment of paraneoplastic pemphigus in follicular NHL with rituximab : report of a case and review of treatment for paraneoplastic pemphigus in NHL and CLL. Am J Hematol 2001 ; 66(2) : 142-144.
- Marazza G, et al : Incidence de la pemphigoïde bulleuse et du pemphigus en Suisse : une étude prospective de 2 ans. Br J Dermatol 2009 ; 161(4) : 861-868.
- de las Heras MN : Dermatose bulleuse à IgA linéaire de l’enfance : bonne réponse au traitement antibiotique. Clin Exp Dermatol 2014 ; 39(3) : 395-397.
- Tashima S, et al : A case of vancomycin-induced linear IgA bullous dermatosis with circulating IgA antibodies to the NC16a domain of BP180. Int J Dermatol 2014 ; 53(3) : 207-209.
- Hundorfean G, Neurath MF, Sitaru C : Autoimmunité contre le collagène de type VII dans la maladie inflammatoire du côlon. J Cell Mol Med 2010 ; 14(10) : 2393-2403.
- Calabresi V, et al : Sensibilité de différents tests pour le diagnostic sérologique de l’épidermolyse bulleuse acquise : analyse d’une cohorte de 24 patients italiens. J Eur Acad Dermatol Venereol 2014 ; 28(4) : 483-490.
- Hall MA, Lanchbury JS, Ciclitira PJ : Gènes de la région HLA classe II et susceptibilité à la dermatite herpétiforme : les associations DPB1 et TAP2 sont secondaires à celles de la sous-région DQ. Eur J Immunogenet 1996 ; 23(4) : 285-296.
- Hervonen K, et al : Dermatitis herpetiformis in children : a long-term follow-up study. Br J Dermatol 2014 [Epub ahead of print].
CONCLUSION POUR LA PRATIQUE
- Pour toutes les dermatoses bulleuses auto-immunes, outre l’anamnèse, l’examen de l’ensemble du tégument, y compris la peau, est indispensable. l’inspection des muqueuses et des ongles est obligatoire.
- Si une maladie bulleuse auto-immune est suspectée cliniquement ou histologiquement, le diagnostic doit être confirmé par la détection des auto-anticorps sous-jacents (coloration des anticorps liés aux tissus en immunofluorescence directe sur une coupe de tissu ou détection dans le sérum par immunofluorescence indirecte ou ELISA).
- Dans le traitement des dermatoses bulleuses auto-immunes, les immunosuppresseurs classiques sont toujours utilisés (par ex. corticostéroïdes, azathioprine).
- Les options thérapeutiques les plus récentes sont les anticorps anti-CD20 (rituximab), qui entraînent une réduction des auto-anticorps.
DERMATOLOGIE PRATIQUE 2014 ; 24(4) : 6-10