En cas d’épaississement ou de dilatation inappropriée d’une ou des deux cavités cardiaques, le cœur n’est plus aussi performant. Les cardiomyopathies se caractérisent par un grand nombre de causes différentes de telles modifications du tissu musculaire cardiaque. Cependant, la distinction entre les différentes manifestations cliniques a un impact important sur le pronostic et les régimes de traitement possibles.
Les cardiomyopathies ont de nombreux visages. En principe, elles peuvent être classées en quatre phénotypes morphologiques, à savoir la cardiomyopathie hypertrophique (CMH), la cardiomyopathie dilatée (CMD), la cardiomyopathie arythmogène (CMAR) et la cardiomyopathie restrictive (CMR). Cependant, cette classification grossière, basée sur la vue, ne correspond pas vraiment aux causes réelles, comme le montre le nombre de sous-catégories. Le Prof. Benjamin Meder, Heidelberg (D), a montré que l’imagerie seule n’est donc pas suffisante pour appréhender la maladie. En conséquence, les caractéristiques fonctionnelles devraient également être prises en compte. En effet, la cardiomyopathie est finalement la description d’un phénotype myocardique morphologique et fonctionnel qui ne peut pas être expliqué par une maladie coronarienne ou des propriétés de remplissage modifiées en raison d’une hypertension artérielle, d’une valvulopathie ou d’une cardiopathie congénitale.
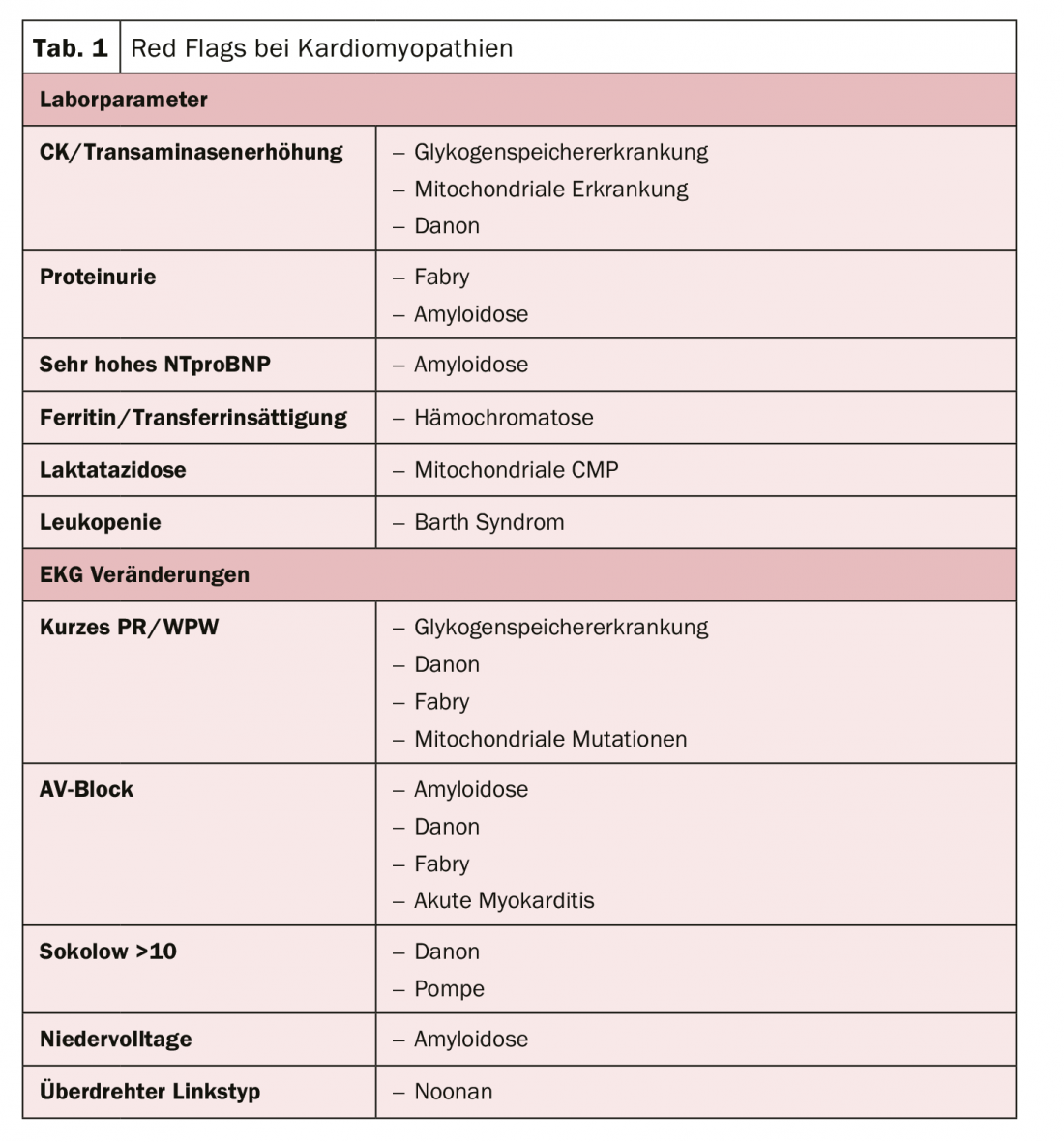
L’approche structurelle d’un traitement optimisé se rapproche donc de l’étiologie dans le but d’éviter la mort subite cardiaque, le problème limitant de toutes les cardiomyopathies. Il existe des paramètres de laboratoire typiques et des anomalies à l’ECG qui peuvent indiquer la présence d’une cardiomyopathie (tableau 1). Il est donc pertinent de pouvoir faire ces distinctions, car le pronostic est essentiellement déterminé par la cause, selon l’expert. Les patients atteints d’amylose, en particulier, présentent une très mauvaise survie.
Détecter l’amylose et la traiter efficacement
L’amylose peut être divisée en trois types en fonction de l’atteinte des organes : L’amylose AL, qui affecte les reins, le cœur et/ou les intestins, l’amylose mt-ATTR, qui affecte principalement le cœur ou le système nerveux, et l’amylose wt-ATTR, qui affecte principalement le cœur. En principe, l’amylose AL peut être considérée comme une complication d’une dyscrasie plasmocytaire. C’est pourquoi un traitement hématologique similaire à celui du myélome multiple est utilisé dans le but d’éliminer le plus rapidement possible les chaînes légères amyloïdogènes (cardio)toxiques. Outre une chimiothérapie à haute dose pour les patients en bonne santé, on utilise principalement un régime à base d’inhibiteurs de protéase. Chez les patients plus jeunes, CyBorD (bortézomib, cyclophosphamide, dexaméthasone) doit être privilégié par rapport à BMDex (bortézomib, melphalan, dexaméthasone) en raison de la toxicité du melphalan sur les cellules souches, afin de préserver la possibilité d’une aphérèse des cellules souches et d’une chimiothérapie à haute dose ultérieures.
Dans le cas d’une amylose causée par des dépôts de protéine transthyrétine, des médicaments comme le Diflunisal et le Tafamidis peuvent stabiliser la protéine mutée. En outre, les thérapies géniques qui réduisent la production de transthyrétine (p. ex. patisiran, inotersen) peuvent réduire les effets sur le système nerveux.
Les patients souffrant d’amylose cardiaque AL et ATTR symptomatique sont en principe soumis aux mêmes recommandations thérapeutiques générales que les patients souffrant d’insuffisance cardiaque. Cependant, même de faibles doses de β-bloquants ou d’IEC peuvent entraîner une hypotension symptomatique. Par conséquent, le traitement des patients atteints d’amylose cardiaque repose en premier lieu sur un dosage correct des diurétiques.
Atteinte syndromique de nombreux systèmes d’organes – Maladie de Fabry
La maladie de Fabry est une maladie génétiquement héréditaire qui se manifeste généralement entre 20 et 40 ans. Une enzyme α-Gal A mal repliée ou non fonctionnelle est formée, ce qui perturbe le transport du réticulum endoplasmique vers le lysosome. Il en résulte une accumulation de substrats lysosomaux. Outre le cœur, les reins, le système nerveux central, mais aussi le système nerveux périphérique et la peau peuvent être impliqués. Si elle n’est pas traitée, l’insuffisance rénale est la cause la plus fréquente de décès chez ces patients, a expliqué le professeur Ingrid Kindermann, Homburg/Saar (D). Plus de la moitié des patients atteints de la maladie de Fabry présentent des symptômes cardiaques. Les arythmies malignes sont les principales causes de mort cardiaque subite.
Pendant longtemps, le traitement s’est limité à un simple soulagement des symptômes. Il existe désormais une option de traitement spécifique qui consiste à remplacer l’enzyme manquante par une enzyme produite par biotechnologie. L’alpha-galactosidase A, produite en laboratoire, est administrée au patient par perfusion et assure la dégradation du matériel de stockage accumulé. Cependant, comme l’enzymothérapie substitutive ne peut pas réparer les dommages déjà causés aux organes, mais seulement retarder leur progression, elle doit être mise en place le plus tôt possible. Une autre possibilité consiste à prendre une chaperonne pharmacologique par voie orale. Cette substance active peut être utilisée chez les patients présentant certaines mutations du gène GLA et une activité résiduelle de l’enzyme α-galactosidase A. En effet, il se lie aux formes instables d’AGAL et stabilise l’enzyme, ce qui lui permet de dégrader les graisses accumulées dans la cellule.
Source : CardioMedLive 2020
CARDIOVASC 2020 ; 19(3) : 28-29 (publié le 17.9.20, ahead of print)