Les dermatoses bulleuses auto-immunes (BAID) sont un groupe de maladies rares, potentiellement mortelles, qui se manifestent cliniquement par des lésions de la peau ou des muqueuses, mais qui, contrairement à l’intuition, ne s’accompagnent pas toujours de la formation de bulles. Les modifications de la peau et des muqueuses sont déclenchées par une réaction du système immunitaire aux protéines structurelles de la peau, médiée par des auto-anticorps.
Les dermatoses bulleuses auto-immunes (BAID) sont un groupe de maladies rares, potentiellement mortelles, qui se manifestent cliniquement par des lésions de la peau ou des muqueuses, mais qui, contrairement à l’intuition, ne s’accompagnent pas toujours de la formation de bulles. Les modifications de la peau et des muqueuses sont déclenchées par une réaction du système immunitaire, médiée par des auto-anticorps (AAK), aux protéines structurelles de la peau qui assurent l’intégrité de l’épithélium lui-même (desmosomes) ou celle de l’ancrage épithélial au tissu conjonctif sous-jacent de la peau ou des muqueuses. Les premières sont regroupées sous le nom de pemphigus et les secondes sous celui de pemphigoïdes (tableau 1) [1,2]. Il faut la distinguer de la dermatite herpétiforme, qui produit des anticorps contre les transglutaminases 2 et 3 et qui est toujours associée à une maladie cœliaque [3].
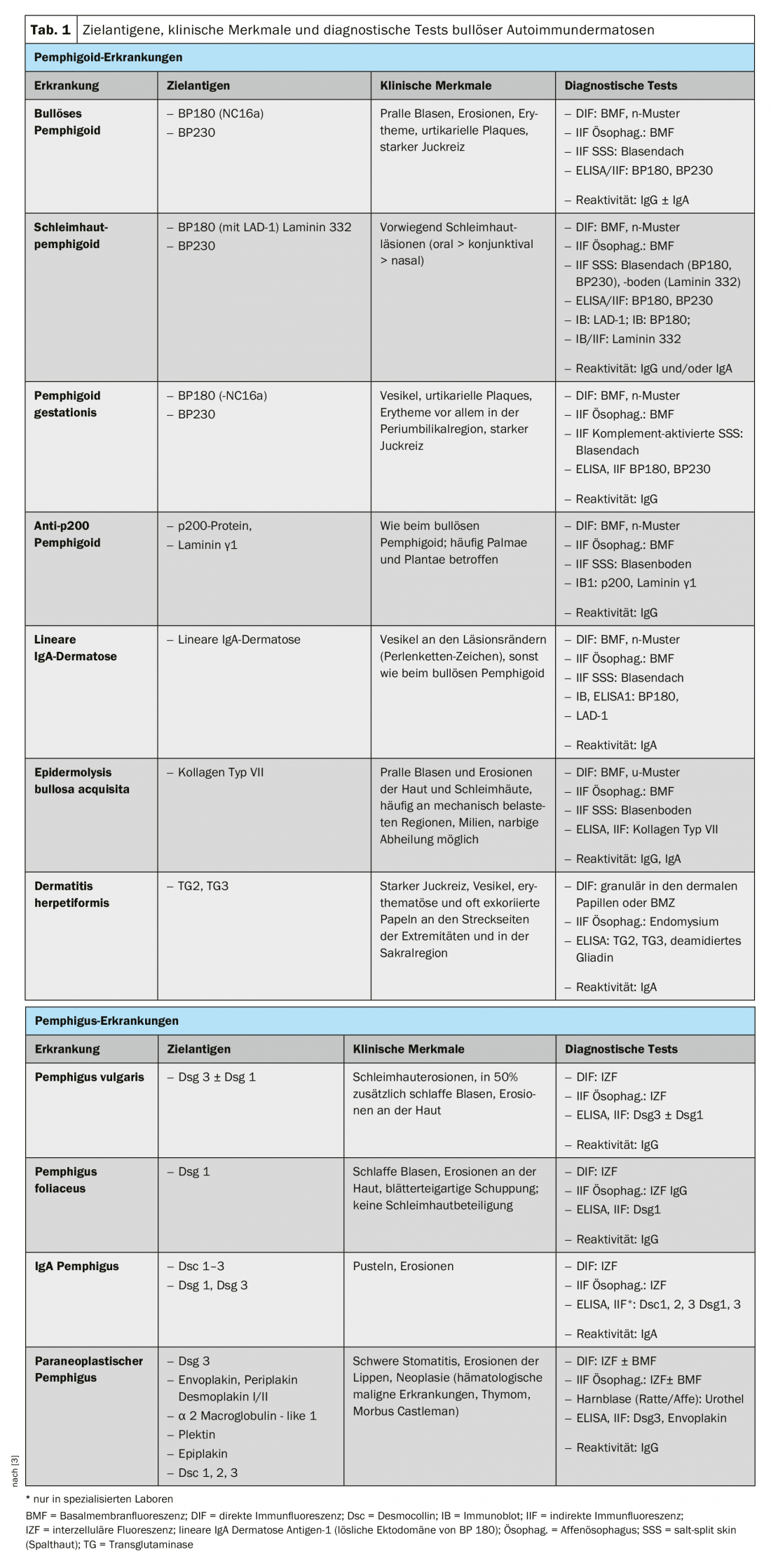
L’antigène cible des auto-anticorps a une influence déterminante sur la pathogenèse et donc sur l’aspect clinique du BAID, sert à établir un diagnostic précis et a une influence décisive sur le pronostic et le traitement à appliquer. Dans ce qui suit, nous nous intéresserons principalement à la reconnaissance et à la différenciation des pemphigus et des pemphigoïdes ainsi qu’aux étapes du diagnostic. Des informations plus détaillées sont disponibles dans les lignes directrices actuelles [4,5] (lignes directrices de l’EADV sur le pemphigus et la DH) ainsi que dans les articles de recherche scientifique et les synthèses publiés régulièrement.
Épidémiologie : à l’exception de la pemphigoïde gestationnelle, toutes les BAID peuvent en principe survenir à tout âge, mais il existe une répartition typique par âge des différentes maladies. Alors que la dermatose à IgA linéaire est la plus fréquente chez les enfants et les adolescents, les personnes atteintes de pemphigoïde bulleuse ne développent généralement la maladie qu’après l’âge de 75 ans, les patients atteints de pemphigus étant en moyenne une à deux décennies plus jeunes. [6,7]. Géographiquement, la pemphigoïde bulleuse domine en Europe du Nord, en Europe centrale et en Amérique du Nord, avec une incidence d’environ 20/million d’habitants/an. Dans le sud de l’Europe, en Israël et en Iran, le pemphigus est la BAID la plus fréquente, avec une incidence annuelle de 5 à 15 millions/habitant [1]. En Suisse, l’incidence de la pemphigoïde bulleuse a été évaluée à 12,1/million/an en 2001/2002 et celle du pemphigus vulgaire et foliacé à 0,6/million/an [8]. Toutes les autres BAID sont encore moins fréquentes que le pemphigus.
Autoantigènes et présentation clinique
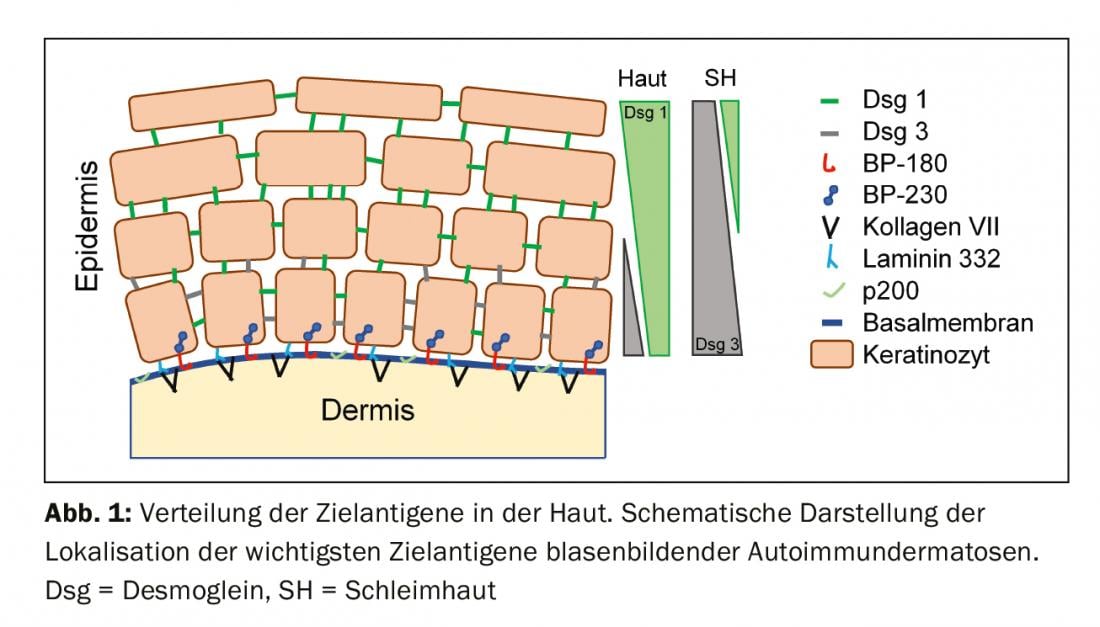
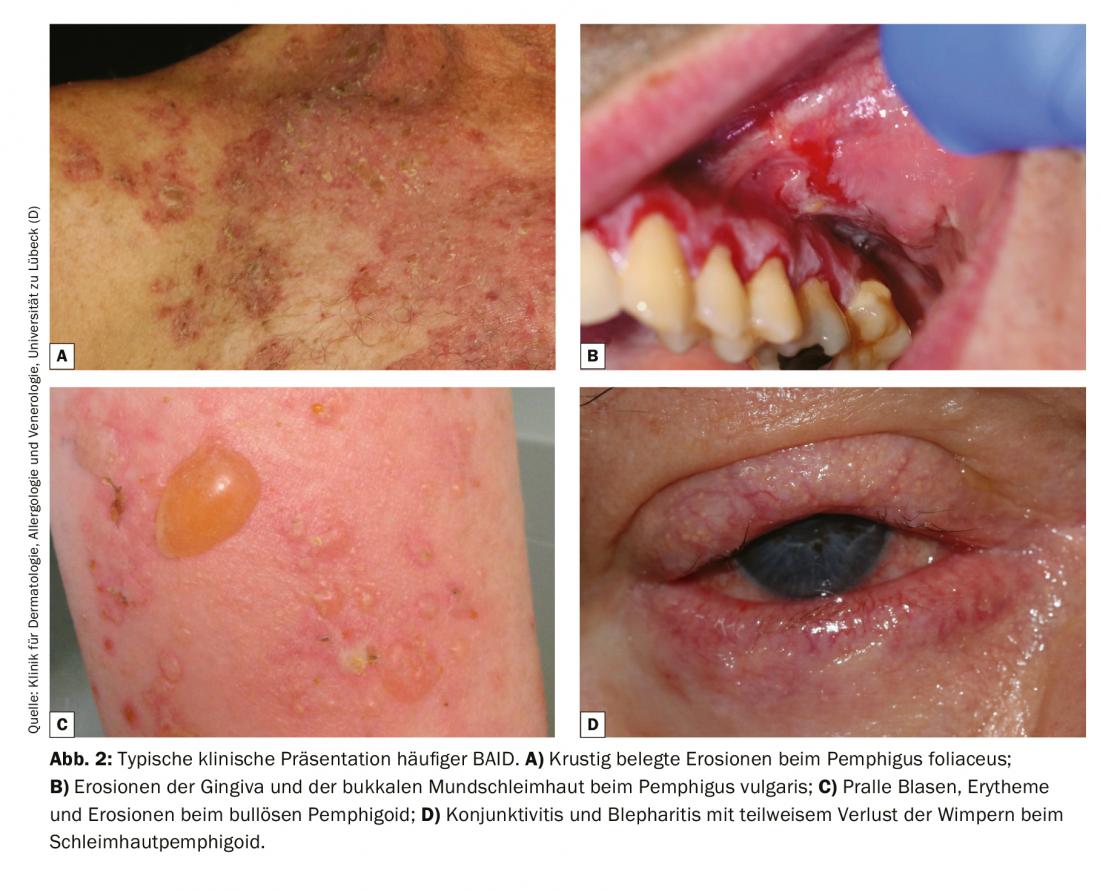
Pour comprendre la clinique d’un BAID, il est utile de regarder d’abord la répartition des antigènes cibles au sein de la peau et des muqueuses (Fig. 1). Les antigènes cibles du groupe pemphigoïde assurent l’ancrage de l’épithélium à la membrane basale et au tissu conjonctif sous-jacent. Cette zone est également appelée zone de jonction dermo-épidermique. Lorsque les auto-anticorps se lient à ces protéines structurelles, une réaction inflammatoire complexe se produit, avec une activation locale du système du complément, une infiltration d’éosinophiles, de neutrophiles, de macrophages et de lymphocytes dans le derme supérieur et, par la suite, la libération d’espèces réactives de l’oxygène et de protéases par ces cellules, ce qui détruit finalement la zone de jonction dermo-épidermique [9]. Il en résulte la formation d’un clivage sous-épidermique et de bulles rebondies (séreuses/hémorragiques) typiques sur le plan clinique (fig. 2C). L’irritation mécanique provoque des érosions de la peau et parfois des muqueuses. Les auto-anticorps de la pemphigoïde bulleuse se lient au BP180 (collagène de type XVII), principalement au domaine BP180 NC16A, ainsi qu’au BP230 chez la moitié des patients (tableau 1). 20% des cas de pemphigoïde bulleuse évoluent sans formation de bulles, avec des tableaux cliniques variés comprenant des érythèmes urticariens, des papules excoriées et de l’eczéma [10]. Il y a pratiquement toujours des démangeaisons prononcées. L’association du BP avec diverses maladies neurologiques chez 30 à 50% des patients, ainsi qu’avec le diabète sucré et les hémopathies malignes, est d’une grande importance clinique [2,11]. On sait depuis peu que certains antidiabétiques oraux, les inhibiteurs de la dipeptidyl peptidase IV, en particulier la vildagliptine, peuvent déclencher un BP, de sorte que ces médicaments ne doivent pas être utilisés en cas de BP [12].
Le domaine NC16A de BP180 est également la région immunodominante dans la pemphigoïde gestationnelle, une BAID qui survient principalement au cours de la seconde moitié de la grossesse et qui guérit généralement 5 à 6 mois après l’accouchement. Toutefois, les récidives sont fréquentes en cas de nouvelle grossesse. La dermatose à IgA linéaire présente des anticorps IgA dirigés contre des épitopes de BP180 situés plus loin en C-terminal.
La pemphigoïde des muqueuses affecte principalement les muqueuses, notamment la cavité buccale et les conjonctives, ainsi que les muqueuses du nez, du pharynx, de la gorge, de la trachée, de l’œsophage et des organes génitaux (fig. 2D). La formation de cicatrices est fréquente, sauf dans la bouche. Les complications de cette cicatrisation sont une éventuelle cécité et une obstruction des voies respiratoires. Dans la pemphigoïde des muqueuses, les autoanticorps sont dirigés contre les épitopes C-terminaux de la BP180 ou de la laminine 332 (tableau 1). La pemphigoïde anti-p200 ressemble cliniquement à la BP, mais les autoanticorps sont dirigés contre une protéine de 200 kDa de la jonction dermo-épidermique. Chez la grande majorité des patients, les auto-anticorps réagissent également avec la laminine γ1 [13]. L’épidermolyse bulleuse acquise est définie par des anticorps dirigés contre le collagène de type VII et se manifeste cliniquement sous deux formes principales, la variante inflammatoire ressemblant à une BP ou à une pemphigoïde muqueuse, ou la variante mécano-bulleuse classique avec des bulles, des érosions et des cicatrices sur les parties du corps exposées comme le dos des mains [14].
Les antigènes cibles du groupe pemphigus, les cadhérines desmosomales Desmoglein 1 et 3 (Dsg 1, 3), sont produits par les cellules épithéliales afin de se lier solidement entre elles (Fig. 1). Lorsqu’elles sont attaquées, l’association de cellules épithéliales se désagrège (acantholyse). Cliniquement, cela peut être démontré en décollant superficiellement, par des forces de cisaillement, une peau périlésionnelle d’apparence saine (phénomène de Nikolski). Cependant, comme la stabilité d’une bulle dépend du degré d’intégrité intraépithéliale et que celle-ci est directement endommagée, on observe dans le pemphigus des bulles flasques qui se rompent rapidement, de sorte que des érosions se forment généralement. La Dsg 1, l’antigène cible du pemphigus foliacé (PF), est principalement produite dans les couches supérieures de l’épiderme de la peau et des muqueuses, où la Dsg 3 n’est pas exprimée, de sorte que le PF se manifeste par des érosions, des croûtes et une desquamation finement lamellaire sur la peau, mais les muqueuses sont toujours exemptes (hypothèse de compensation de la Dsg), Fig. 2A) [1,15]. Dsg 3, l’antigène cible du pemphigus vulgaire (PV), est principalement exprimé dans les couches inférieures de l’épithélium ; toutefois, à ces endroits, Dsg1 n’est présent que dans la muqueuse épithéliale et non dans la peau. Ainsi, en présence d’anticorps anti-Dsg3 uniquement, la PV provoque exclusivement des érosions de la muqueuse (fig. 2B), qui sont souvent douloureuses, gênent l’alimentation et peuvent entraîner une perte de poids. Dans le cas de la PV muco-cutanée avec anticorps anti-Dsg3 et anti-Dsg1, des érosions et des bulles flasques apparaissent sur la peau en plus des lésions des muqueuses. Contrairement aux pemphigoïdes, aucune réaction inflammatoire n’est nécessaire pour la formation de bulles ; la seule liaison des anticorps anti-Dsg aux cellules épithéliales est suffisante pour déclencher une acantholyse. L’empêchement stérique de l’interaction desmosomale par la liaison des auto-anticorps, une modification de l’expression des molécules Dsg à la surface des cellules ainsi qu’un remaniement complexe du cytosquelette des cellules épithéliales médié par la transduction du signal jouent ici un rôle décisif [15]. Le pemphigus paranéoplasique est toujours associé à une néoplasie, généralement un thymome ou une hémopathie maligne, et se caractérise cliniquement par une stomatite marquée et une atteinte fréquente des lèvres. Outre la Dsg 3, les auto-anticorps sont dirigés contre les protéines de la famille des plakines (notamment Envoplakin, Periplakin, BP230) et contre l’α2-Macroglobulin-like 1 [1].
Diagnostic
Étant donné que la présentation clinique du BAID est si variée et ne peut pas toujours être clairement attribuée à un BAID spécifique, le diagnostic différentiel doit exclure les causes infectieuses (impétigo, érysipèle bulleux, syndrome de la peau échauffée par les staphylocoques, infections par le virus de l’herpès), les maladies héréditaires (porphyrie, épidermolyse bulleuse héréditaire) ainsi que les exanthèmes médicamenteux et les toxines chimiques et physiques [4]. En cas d’atteinte de la cavité buccale, le diagnostic différentiel doit surtout porter sur un lichen ruber mucosae, et en cas d’atteinte oculaire, sur une origine infectieuse, allergique ou irritative. En particulier en cas d’atteinte oculaire, il faut penser rapidement à une pemphigoïde des muqueuses, car c’est le seul moyen d’éviter une cicatrisation irréversible sur le plan thérapeutique. La figure 2 et le tableau 1 résument les caractéristiques cliniques.
En plus de l’examen clinique, l’immunofluorescence directe (IF) d’une biopsie périlésionnelle et la détection des autoanticorps circulants par IF indirecte, ELISA (Enzyme-linked Immunosorbent Assay) et immunoblots sont essentiels pour le diagnostic du BAID [4].
Le diagnostic exact de chaque BAID est à la fois important pour le pronostic et pour la thérapie. Ainsi, le pemphigus paranéoplasique est pratiquement toujours associé à une tumeur maligne et la pemphigoïde muqueuse antilaminine-332 est associée à une tumeur maligne dans 25-30% des cas et une recherche de tumeur est indiquée chez ces patients [1,16,17]. Les maladies pemphigus, l’épidermolyse bulleuse acquise et la pemphigoïde muqueuse avec atteinte oculaire, laryngée ou trachéale ne peuvent être traitées que par un traitement immunosuppresseur intensif, tandis que la pemphigoïde anti-p200, les dermatoses à IgA linéaire et la dermatite herpétiforme ne nécessitent généralement qu’une immunosuppression légère ou un régime sans gluten.
Immunofluorescence directe
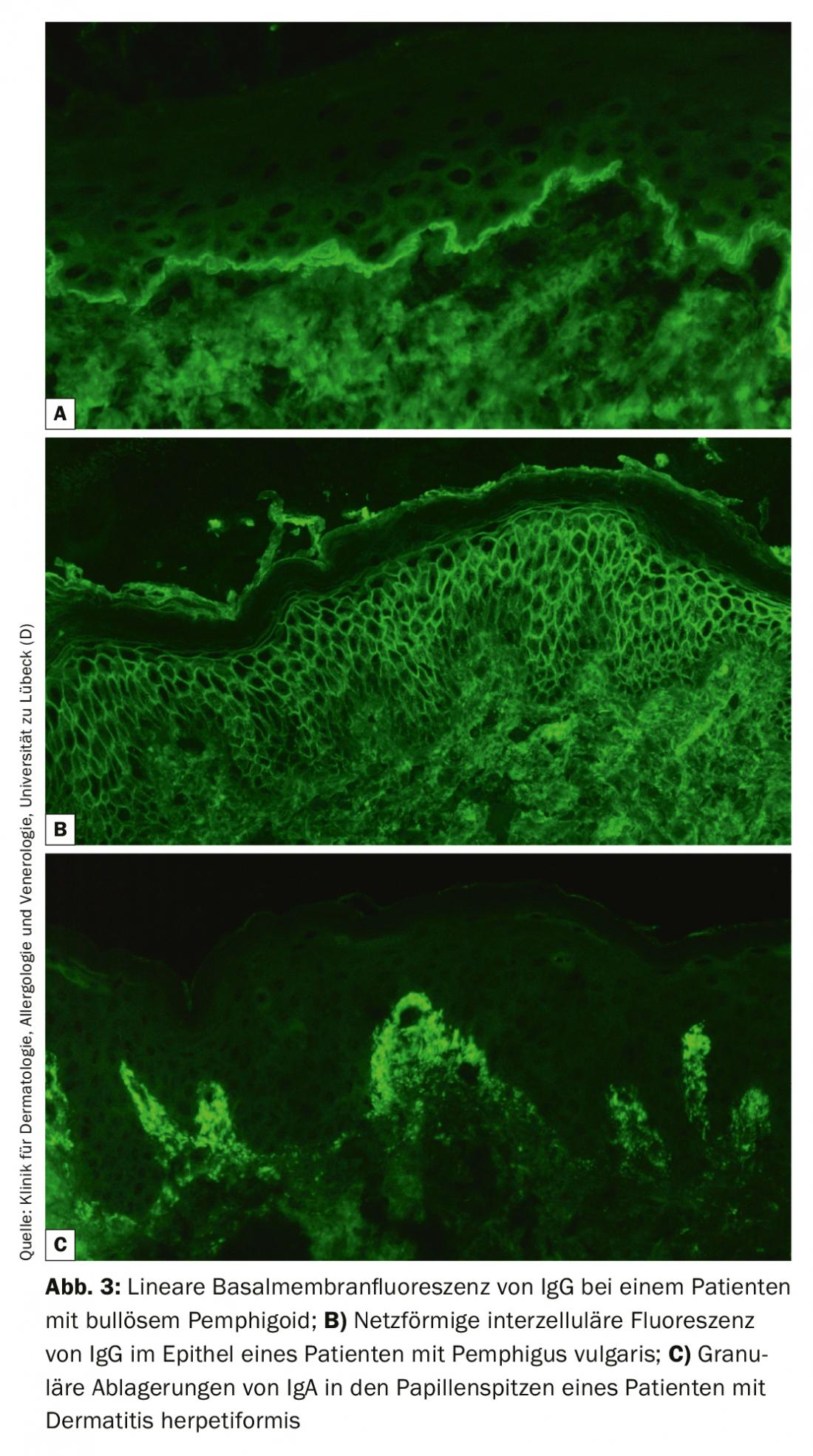
La FI directe d’une biopsie périlésionnelle reste le gold standard diagnostique du BAID avec une spécificité de 98% et une sensibilité de 80-90% [4]. Une fois prélevée, la biopsie de l’échantillon doit être immédiatement congelée ou stockée dans du NaCl 0,9% ou du milieu de Michels et traitée dans les 72 heures. Les autoanticorps liés aux tissus (immunoglobulines [Ig] G, A et M) et le facteur C3 du complément sont ensuite mis en évidence sur des coupes congelées à l’aide d’un anticorps marqué par fluorescence. La reconnaissance d’un motif de fluorescence typique permet en principe de diagnostiquer avec certitude un pemphigus (fluorescence intercellulaire épithéliale/motif réticulaire), une pemphigoïde (fluorescence linéaire le long de la membrane basale) et une dermatite herpétiforme (dépôts granuleux d’IgA dans les pointes papillaires ou le long de la membrane basale, Figure 3). En complément, une biopsie lésionnelle doit être réalisée afin de procéder à un examen histologique ; celui-ci permet de diagnostiquer d’autres maladies, en particulier si l’IF directe et la sérologie sont négatives.
Immunofluorescence indirecte sur les substrats d’organes
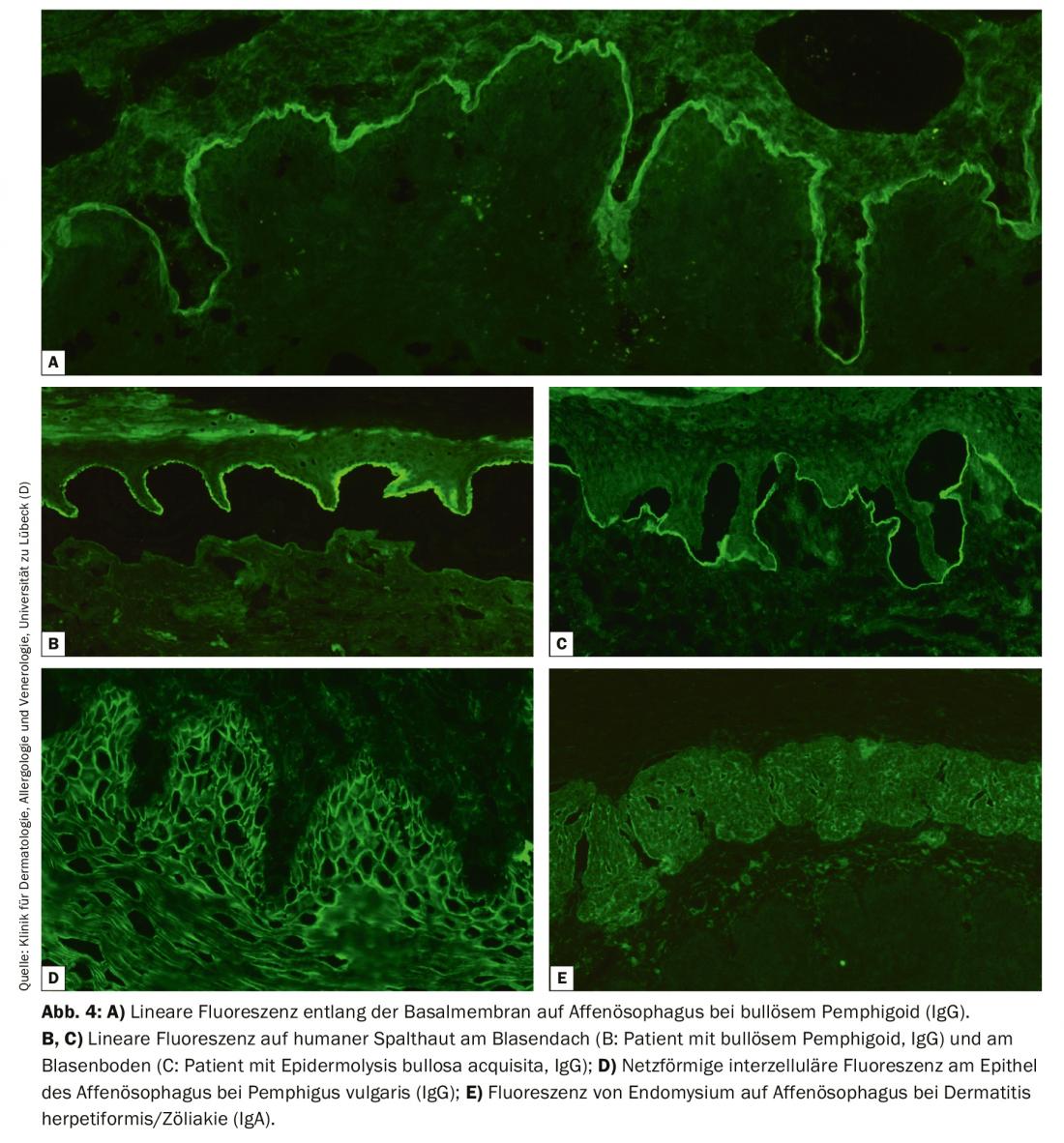
Pour l’IF indirecte, on utilise généralement l’œsophage de primate et du NaCl-Lsg. de la peau humaine fendue au niveau de la membrane basale, appelée peau fendue, est utilisée pour détecter les autoanticorps sériques dans le BAID. La coupe de l’œsophage révèle la présence d’auto-anticorps le long de la membrane basale dans les pemphigoïdes. (Fig. 4A), dans les maladies pemphigus, intercellulaire sur l’épithélium (ill. 4D) et dans la dermatite herpétiforme et la maladie cœliaque contre l’endomysium (IgA uniquement, Figure 4E). Dans la peau fendue, les autoanticorps sériques des maladies pemphigoïdes se lient soit au toit de la bulle artificielle (dans le cas du BP, de la pemphigoïde muqueuse anti-BP180, de la pemphigoïde gestationis, de la dermatose linéaire à IgA, Fig. 4B) ou dans le fond de la vessie (dans la pemphigoïde anti-p200, la pemphigoïde muqueuse anti-laminine 332, l’épidermolyse bulleuse acquise, Fig. 4C). Ici aussi, on recherche les dépôts d’IgG et d’IgA, ces derniers étant par exemple typiques de la dermatose à IgA linéaire. L’examen par vessie de singe/de rat dans le pemphigus paranéoplasique ou le test de fixation du complément sur la peau fendue en cas de suspicion de pemphigoïde gestationnelle sont plus rarement indiqués.
L’IF indirecte sur les substrats d’organes mentionnés est utilisée comme test de dépistage, puis, en fonction du résultat, des tests spécifiques à l’antigène, ELISA, IF indirecte, immunoblots ou immunoprécipitation peuvent être utilisés en tenant compte de l’IF directe. Le développement de tests spécifiques à l’antigène, utilisant généralement les formes recombinantes immunodominantes des auto-antigènes, permet aujourd’hui de poser un diagnostic sérologique chez 80 à 90% des patients atteints de BAID [18–20].
ELISA
Des tests ELISA commerciaux très sensibles et spécifiques sont disponibles pour les principaux antigènes cibles, Dsg1, Dsg3, Envoplakin, BP180 NC16A, BP230 et le collagène de type VII [3]. Pour les anticorps anti-Dsg1, BP180 NC16A, collagène de type VII et, dans une moindre mesure, Dsg3, il a été démontré que les taux sériques sont bien corrélés à l’activité clinique sur la peau et les muqueuses des patients atteints de pemphigus, de pemphigoïde bulleuse et d’épidermolyse bulleuse acquise, de sorte que les tests ELISA peuvent également être utilisés pour surveiller l’activité de la maladie au cours de la maladie. Ceci est particulièrement pertinent pour les patients en rémission clinique, chez qui il faut décider dans quelle mesure le traitement immunosuppresseur peut être encore réduit.
Systèmes de tests multivariés : des systèmes de tests multivariés ont été développés afin d’examiner différentes spécificités d’autoanticorps en une seule étape et de gagner du temps par rapport à la procédure en plusieurs étapes présentée ci-dessus. D’une part, un test ELISA dans lequel les protéines recombinantes ont été placées sur une seule plaque ELISA [18], d’autre part, basé sur la technologie Biochip®. Ici, jusqu’à 12 substrats d’environ 1×1 mm seulement sont compilés dans un champ d’incubation sur une lame de laboratoire normale et la liaison des autoanticorps est visualisée par IF indirecte. Outre les substrats d’organes tels que l’œsophage de primate et la peau fendue humaine, le BP180 NC16A recombinant ainsi que les Dsg1, Dsg3, BP230, la laminine 332, le collagène de type VII, exprimés à la surface des cellules HEK293, sont disponibles [19].
Immunoblot et immunoprécipitation
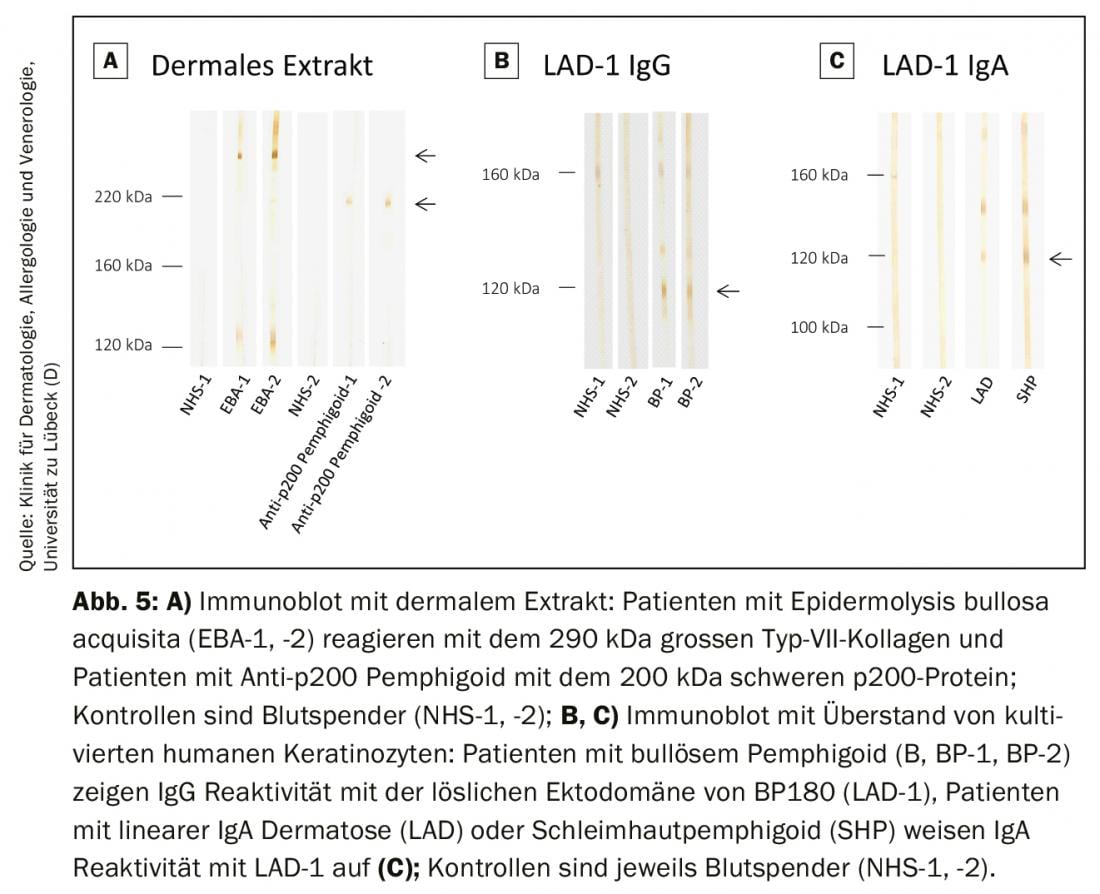
Dans les laboratoires spécialisés, ces méthodes sont proposées en tant que procédures internes pour détecter les spécificités des autoanticorps qui ne sont pas encore couvertes par les systèmes de test commerciaux. Cela inclut la détection d’auto-anticorps contre la molécule complète de collagène de type VII et la protéine p200. (ill. 5A) et la détection d’anticorps IgG et IgA dirigés contre l’ectodomaine de BP180, qui est la région immunodominante dans la pemphigoïde des muqueuses et les dermatoses à IgA linéaires. (Fig. 5B et C). Les anticorps contre la périplakine, la desmoplakine I/II, la plectine, l’épiplakine, la desmocolline et l’α2-macroglobuline-like 1 dans le pemphigus paranéoplasique peuvent être visualisés dans différentes analyses d’immunoblot et d’immunoprécipitation. En Allemagne, l’organisme d’accréditation allemand (DAkkS) offre la possibilité d’auditer ces procédures internes en externe afin de garantir un niveau de diagnostic aussi élevé que possible. Le laboratoire auto-immun de la clinique universitaire de dermatologie de Lübeck propose ces procédures (DAkkS D-ML-13069-06-00 ; www.uksh.de/dermatologie-luebeck).
Constellations particulières de résultats
Si les résultats ne sont pas clairs, il est recommandé de prélever à nouveau un échantillon de biopsie périlésionnelle pour la FI directe [4]. Dans le cas de la pemphigoïde des muqueuses, il a été montré que chez 16% des patients, le diagnostic ne pouvait être confirmé que par une seconde IF directe [21]. L’IF directe revêt une importance particulière pour la pemphigoïde des muqueuses et l’épidermolyse bulleuse acquise, car dans 50% des cas, aucun auto-anticorps circulant n’est détecté.
Pour la grande majorité des patients atteints de BAID, le diagnostic peut aujourd’hui déjà être effectué de manière sérologique à l’aide de systèmes de test commerciaux si le tableau clinique est compatible [18,19]. Nous recommandons également les tests sérologiques comme examen de dépistage initial lorsqu’il est préférable de ne pas effectuer de biopsie d’échantillon dans un premier temps, par exemple chez les enfants ou les patients >75 ans avec >6 semaines de démangeaisons qui ne peuvent pas être expliquées par une autre maladie.
Les directives actuelles de l’AWMF [5] recommandent le diagnostic de pemphigus vulgaire/foliacé en cas de
- un tableau clinique approprié et une immunofluorescence directe positive
- un tableau clinique approprié et une réactivité avec la desmogléine 1 ou 3 en ELISA/dans des cellules transfectées
- tableau clinique et histopathologie appropriés et immunofluorescence indirecte positive sur des coupes d’œsophage de singe.
Diagnostic d’une pemphigoïde bulleuse en cas de
- tableau clinique approprié et IF directe positive et réactivité avec BP180 et/ou BP 230
- un tableau clinique approprié et une IF directe positive et une liaison épidermique des IgG dans l’IF indirecte sur la peau fendue
- un tableau clinique avec des bulles rebondies et une fixation épidermique des IgG en IF indirecte sur la peau fendue ou sur des coupes d’œsophage de singe et une réactivité avec BP180 et/ou BP230
- clinique avec bulles rebondies et histopathologie correspondante et fixation épidermique d’IgG dans l’IF indirecte sur peau fendue
- tableau clinique et histopathologie appropriés (clivage sous-épidermique) et réactivité avec BP180
- un tableau clinique avec des bulles rebondies et une réactivité évidente avec le BP180 (par exemple >3 fois la limite inférieure de détection dans le test ELISA commercial)
Résumé
Les dermatoses bulleuses auto-immunes (BAID) sont un groupe de maladies rares, potentiellement mortelles, qui se manifestent cliniquement par des lésions de la peau ou des muqueuses et qui ne sont pas toujours associées à la formation de bulles. Les modifications de la peau et des muqueuses sont déclenchées par une réaction du système immunitaire, médiée par des auto-anticorps, à des protéines structurelles de la peau qui assurent l’intégrité de l’épithélium lui-même (desmosomes) ou celle de l’ancrage épithélial au tissu conjonctif sous-jacent de la peau ou des muqueuses. Les premières sont regroupées sous le nom de pemphigus et les secondes sous celui de pemphigoïdes. Il faut la distinguer de la dermatite herpétiforme, dans laquelle des anticorps contre la transglutaminase 2 et 3 sont produits et qui est toujours associée à une maladie cœliaque. L’antigène cible des autoanticorps a une influence déterminante sur la pathogenèse et donc sur l’aspect clinique du BAID. Sa détermination ne sert pas seulement à établir un diagnostic précis, mais a une influence décisive sur le pronostic et le traitement à appliquer. Le diagnostic du BAID repose sur la détection d’autoanticorps liés aux tissus en immunofluorescence directe sur une biopsie d’échantillon périlésionnel et sur la détection d’autoanticorps circulants. Connaissant le tableau clinique, les tests ELISA et d’immunofluorescence indirecte modernes, basés sur les régions immmunodominantes des antigènes cibles, permettent déjà de diagnostiquer sérologiquement 80 à 90% des patients atteints de BAID. Le développement de systèmes de tests sensibles et spécifiques standardisés supplémentaires permettra de simplifier davantage le diagnostic de la BAID.
Messages Take-Home
- Les dermatoses bulleuses auto-immunes sont des maladies rares provoquées par des auto-anticorps dirigés contre des protéines structurelles de la peau.
- La pemphigoïde bulleuse est de loin la dermatose auto-immune la plus fréquente en Europe du Nord et en Europe centrale. Elle se caractérise cliniquement par un âge avancé, un prurit intense, des bulles rebondies et une association avec des troubles neurologiques.
- L’établissement d’un diagnostic précis a une importance pronostique et thérapeutique.
- Des tests ELISA standardisés, hautement sensibles et spécifiques, et des tests d’immunofluorescence indirecte utilisant la région immunodominante des antigènes cibles sont disponibles.
- Connaissant le tableau clinique, les tests sérologiques permettent un diagnostic de certitude dans la grande majorité des patients, la biopsie périlésionnelle de l’échantillon pour un examen direct par immunofluorescence reste l’étalon-or du diagnostic.
Remerciements
Nous remercions Ingeborg Atefi et Marina Kongsbak-Reim pour leur aide dans la réalisation des images de fluorescence, ainsi que Vanessa Krull pour son aide dans la réalisation des immunoblots. Ce travail a été soutenu par un financement structurel du cluster d’excellence 2167/1 Precision Medicine in Chronic Inflammation.
Littérature :
- Schmidt E, Kasperkiewicz M, Joly P : Pemphigus. Lancet 2019 ; 394 : 882-894.
- Schmidt E, Zillikens D : Maladies pemphigoïdes. Lancet 2013 ; 381 : 320-332.
- van Beek N, Zillikens D, Schmidt E : Diagnostic des maladies bulleuses auto-immunes. J Dtsch Dermatol Ges 2018 ; 16 : 1077-1091.
- Schmidt E, Goebeler M, Hertl M, et al : S2k guideline for the diagnosis of pemphigus vulgaris/foliaceus and bullous pemphigoid. J Dtsch Dermatol Ges 2015 ; 13 : 713-727.
- Schmidt E, Sticherling M, Sardy M, et al : S2k guidelines for the treatment of pemphigus vulgaris/foliaceus and bullous pemphigoid : 2019 update. J Dtsch Dermatol Ges 2020 ; 18 : 516-526.
- Hübner F, Recke A, Zillikens D, et al : Prevalence and Age Distribution of Pemphigus and Pemphigoid Diseases in Germany. The Journal of investigative dermatology (JEADV) 2016 : 136(12) : 2495-2498 ; doi : 10.1016/j.jid.2016.07.013.
- Hübner F, König IR, Holtsche MM, et al : Prevalence and age distribution of pemphigus and pemphigoid diseases among paediatric patients in Germany. In : JEADV 2020 ; doi : 10.1111/jdv.16467.
- Marazza G, Pham HC, Scharer L, et al : Incidence de la pemphigoïde bulleuse et du pemphigus en Suisse : une étude prospective de 2 ans. Br J Dermatol 2009 ; 161 : 861-868.
- Sadik CD, Schmidt E : Résolution de la pemphigoïde bulleuse. Seminars in immunopathology 2019 ; 41 : 645-654.
- della Torre R, Combescure C, Cortes B, et al : Présentation clinique et retard de diagnostic dans la pemphigoïde bulleuse : une cohorte nationale prospective. Br J Dermatol 2012 ; 167 : 1111-1117.
- Schulze F, Neumann K, Recke A, et al : Malignancies in pemphigus and pemphigoid diseases. J Invest Dermatol 2015 ; 135 : 1445-1447.
- Kridin K, Cohen AD : Pemphigoïde bulleuse associée à l’inhibiteur de la dipeptidyl-peptidase IV : une revue systématique et une méta-analyse. J Am Acad Dermatol 2018.
- Goletz S, Hashimoto T, Zillikens D, Schmidt E : Pemphigoïde anti-p200. J Am Acad Dermatol 2014 ; 71 : 185-191.
- Vorobyev A, Ludwig RJ, Schmidt E : Caractéristiques cliniques et diagnostic de l’épidermolyse bulleuse acquise. Expert Rev Clin Immunol 2017 ; 13 : 157-169.
- Kasperkiewicz M, Ellebrecht CT, Takahashi H, et al : Pemphigus. Nature reviews Disease primers 2017 ; 3 : 17026.
- Egan CA, Lazarova Z, Darling TN, et al : Anti-epiligrin cicatricial pemphigoid and relative risk for cancer. Lancet 2001 ; 357 : 1850-1851.
- Goletz S, Probst C, Komorowski L, et al : A sensitive and specific assay for the serological diagnosis of antilaminin 332 mucous membrane pemphigoid. Br J Dermatol 2019 ; 180 : 149-156.
- van Beek N, Dahnrich C, Johannsen N, et al : Prospective studies on the routine use of a novel multivariant enzyme-linked immunosorbent assay for the diagnosis of autoimmune bullous diseases. J Am Acad Dermatol 2017 ; 76 : 889-894 e5.
- van Beek N, Kruger S, Fuhrmann T, et al : Multicenter prospective study on multivariant diagnosis of autoimmune bullous dermatoses using the BIOCHIP(TM) technology. J Am Acad Dermatol 2020.
- van Beek N, Rentzsch K, Probst C, et al : Serological diagnosis of autoimmune bullous skin diseases : Prospective comparison of the BIOCHIP mosaic-based indirect immunofluorescence technique with the conventional multi-step single test strategy. Orphanet J Rare Dis 2012 ; 7 : 49.
- Shimanovich I, Nitz JM, Zillikens D : Des prélèvements multiples et répétés augmentent la sensibilité du test d’immunofluorescence directe pour le diagnostic de la pemphigoïde à membranes muqueuses. J Am Acad Dermatol 2017 ; 77 : 700-705 e3.
DERMATOLOGIE PRATIQUE 2020 ; 30(5) : 12-19