Les antihistaminiques H1 sont divisés en une première génération, plus ancienne, avec des effets secondaires sédatifs et une deuxième génération sans ces effets. Notre auteur met en lumière la pharmacologie de ce groupe de substances actives.
Jusqu’à présent, quatre récepteurs de l’histamine différents ont été identifiés. Ces molécules couplées à des protéines G sont situées à la surface des cellules et exercent des effets différents selon leur lieu d’expression. Alors que l’activation du récepteur H1 entraîne notamment un prurit, une vasodilatation, une contraction des muscles lisses avec bronchospasme ou crampes abdominales, une sécrétion de mucosités avec rhinorrhée et augmentation des sécrétions bronchiques ainsi qu’une augmentation de la perméabilité vasculaire, les récepteurs H2 sont notamment impliqués dans l’augmentation de la sécrétion de sucs et d’acides gastriques. Il existe également des récepteurs H3, qui jouent un rôle dans le SNC en tant qu’autorécepteurs présynaptiques, et des récepteurs H4, qui jouent notamment un rôle dans la différenciation et la modulation des cellules immunitaires. Dans cet article, les substances dirigées contre les récepteurs H1 seront abordées, tandis que les antagonistes des récepteurs H2, dont seule la ranitidine est encore commercialisée en Suisse, ne seront pas traités. Les agonistes et antagonistes des récepteurs H3 et H4 sont en cours de développement clinique.
Pharmacodynamie des antihistaminiques H1
En Suisse, 22 substances actives de la classe des antihistaminiques H1 sont disponibles. On pense aujourd’hui que les antihistaminiques stabilisent le récepteur H1 dans sa conformation inactive, ce qui permet de réduire le nombre de récepteurs pouvant être activés par l’histamine. Alors que les antihistaminiques H1 de la première génération, plus ancienne, pénètrent bien dans le système nerveux central et y exercent un effet sédatif sur les récepteurs H1 postsynaptiques, ce n’est pas le cas des représentants de la 2e génération ou alors à des concentrations thérapeutiques (tableau 1). En raison de leur bonne efficacité sur les récepteurs H1 centraux, certains représentants de la génération 2 sont utilisés dans le traitement de la dépression. 1ère génération comme sédatif/hypnotique (p. ex. doxylamine, diphenhydramine), antiémétique (p. ex. méclozine) ou contre le mal des transports (p. ex. dimenhydrinate). La mauvaise pénétration du SNC des représentants de la 2ème génération est due au fait que ces substances sont hydrophiles et sont des substrats du transporteur vers l’extérieur P-glycoprotéine présent dans la barrière hémato-encéphalique (entre autres barrières membranaires de l’organisme). Cela permet d’éviter la sédation utilisée dans les indications antiallergiques avec les substances de la classe 1ère génération était souvent limitante pour le traitement. Certains antihistaminiques H1 de la 1ère génération ont des effets supplémentaires sur les récepteurs de l’acétylcholine, de la noradrénaline et de la sérotonine, tandis que les représentants des 2ème génération inactivent spécifiquement le récepteur H1.
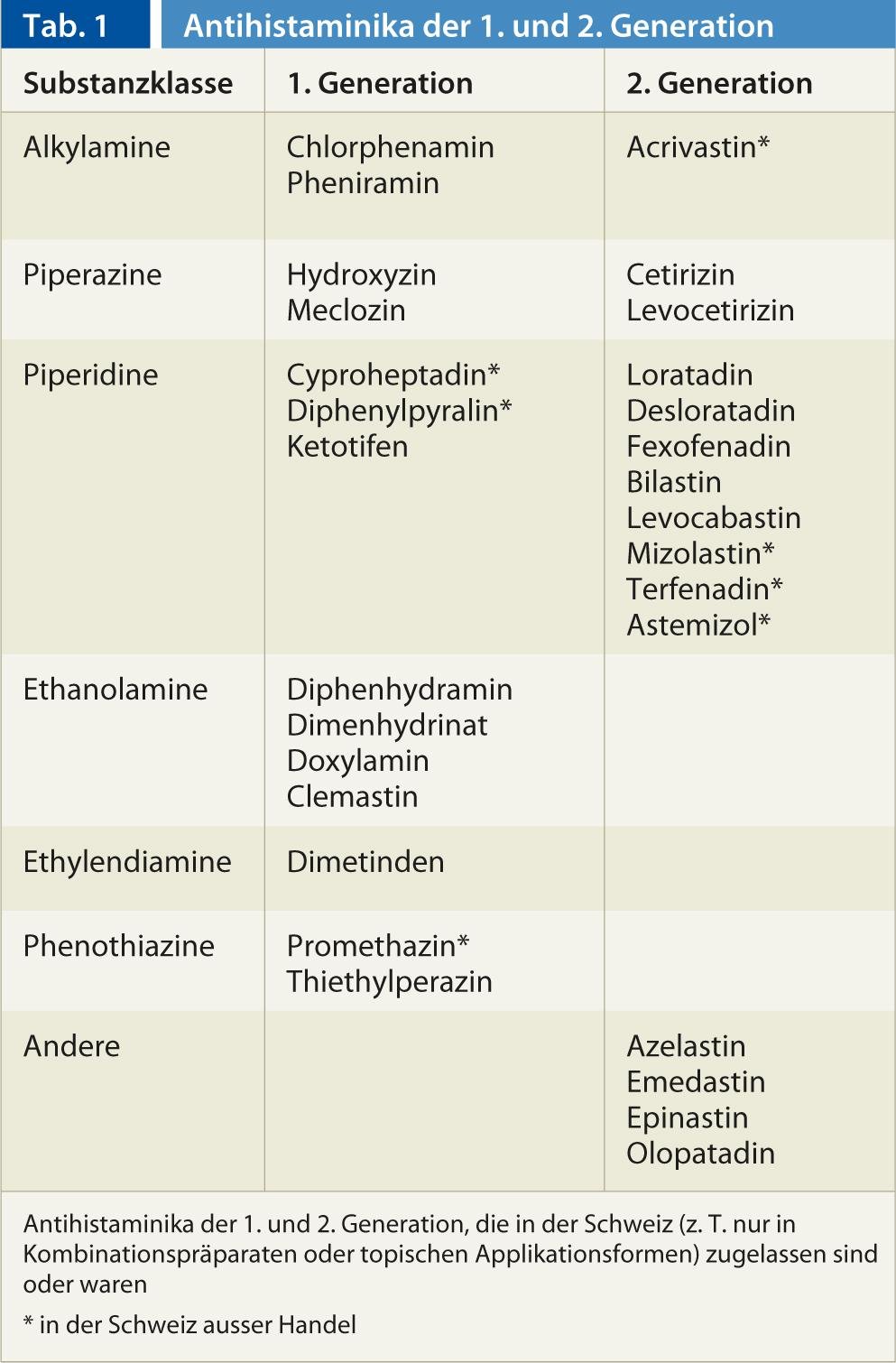
Dans l’ensemble, l’efficacité clinique des antihistaminiques H1 du 1ère génération sont mal étudiés dans les essais cliniques, tandis que les preuves de l’utilisation des antihistaminiques H1 de la 2ème génération dans la rhinite allergique, la conjonctivite allergique et l’urticaire. L’utilisation de produits de 2ème génération dans la dermatite atopique, l’asthme, l’anaphylaxie, l’angio-œdème non allergique, les rhumes, les démangeaisons d’origine non allergique, etc. est mal étudiée dans les études ou les études n’ont pas montré d’effets convaincants et il n’existe pas non plus d’autorisation de mise sur le marché pour de telles indications. Chez les enfants, les substances de la Les médicaments de 1ère génération peuvent entraîner des effets secondaires parfois menaçants, de sorte que l’indication doit être posée avec un soin particulier. Dans une soi-disant 3ème génération, des énantiomères ou des métabolites de molécules de la 2ème génération, sans qu’il y ait de grandes différences pharmacodynamiques, de sorte que les substances sont pharmacologiquement proches de la 2ème génération. 2ème génération.
Pharmacocinétique des antihistaminiques H1
Le premier obstacle qu’un médicament doit franchir pour être efficace est la prise. La garantie de l’observance ou de l’adhésion revêt donc une importance particulière dans la pratique médicale quotidienne. La plupart des antihistaminiques H1 à usage systémique sont disponibles sous forme de médicaments solides (comprimés, dragées, comprimés pelliculés, suppositoires) ou de gouttes à prendre par voie orale. Certaines substances sont appliquées localement (par exemple, des gouttes pour les yeux). Seuls quelques-uns peuvent également être administrés par voie intraveineuse (dimétindène, clémastine, thiéthylperazine). L’absorption, qui est rapide pour la plupart des antihistaminiques H1 de deuxième génération, avec des pics de concentration après une à trois heures, est suivie de la distribution dans le sang et les tissus, éventuellement du métabolisme, et de l’excrétion.
Il existe notamment de grandes différences entre les antihistaminiques en termes de métabolisme et d’élimination (tableau 2). En outre, il existe des différences de métabolisme entre les individus, dues à des influences génétiques et environnementales. En particulier, l’enzyme CYP2D6 du cytochrome P450, qui est impliquée dans la dégradation de certains antihistaminiques H1 (tableau 2), présente une forte variabilité génétique, qui peut aller de l’absence à la multiplication de l’activité enzymatique normale. En outre, le CYP2D6 peut être inhibé par certaines substances : le bupropion, le cinacalcet, les inhibiteurs sélectifs de la recapture de la sérotonine (paroxétine, fluoxétine et duloxétine) et l’antifongique terbinafine comptent parmi les inhibiteurs les plus puissants du CYP2D6. Mais comme tous les antihistaminiques H1 sont dégradés par plus d’une enzyme et sont souvent éliminés par voie rénale sans être modifiés, la variabilité génétique et l’inhibition du CYP2D6 ne jouent que très rarement un rôle dans l’utilisation des antihistaminiques H1.
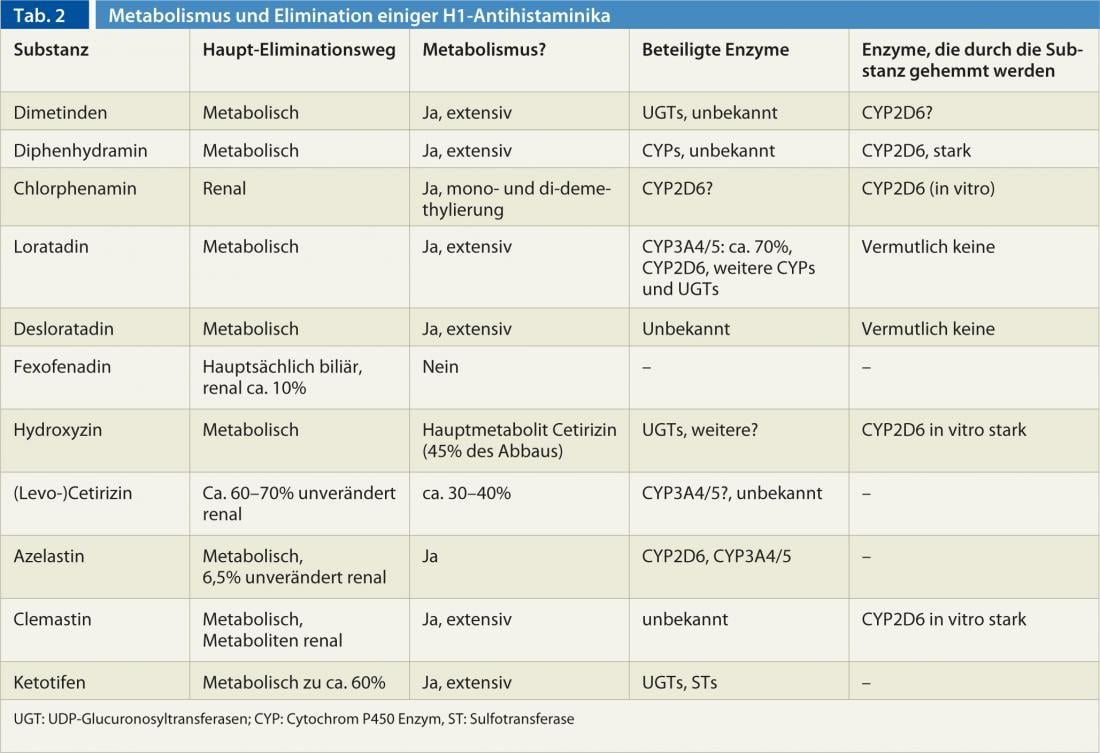
Certains antihistaminiques H1 de première génération sont également des inhibiteurs du CYP2D6. (tableau 2), de sorte qu’en cas d’utilisation concomitante de substrats du CYP2D6 (par ex. codéine, dextrométhorphane, de nombreux antipsychotiques [halopéridol, rispéridone, aripiprazole], atomoxétine, de nombreux antidépresseurs [la plupart des tricycliques, venlafaxine et autres], ainsi que les bêtabloquants métoprolol, carvédilol et timolol), il convient de choisir une dose plus faible du substrat afin d’éviter les effets indésirables.
Dans l’ensemble, la pharmacocinétique des substances de 1. La pharmacologie des médicaments de 1ère génération n’est qu’imparfaitement connue, car les substances ont été autorisées il y a des décennies avec beaucoup moins de données que ce qui est nécessaire aujourd’hui. Mais il existe également des lacunes dans les connaissances sur les préparations de deuxième génération : La desloratadine est métabolisée en un métabolite également actif, la 3-hydroxy-désloratadine, mais la ou les enzymes impliquées sont inconnues, bien qu’il ait été découvert que 2% des Européens et jusqu’à 20% des Africains ne peuvent pas produire de 3-hydroxy-désloratadine. Les enzymes CYP2D6 et CYP3A4/5 impliquées dans le métabolisme de la loratadine ne semblent pas être responsables de ce phénomène.
Le CYP3A4 est la principale enzyme du cytochrome P450, tant en termes de quantité que de nombre de substrats. Alors que des variantes génétiques altérant la fonction du CYP3A4 ne sont pas connues (malgré des recherches intensives), il existe des médicaments qui augmentent l’activité de l’enzyme (rifampicine, éfavirenz, phénytoïne, carbamazépine, ingrédients du millepertuis, etc.) et les inhibent (antifongiques azolés, érythromycine, clarithromycine, ritonavir, vérapamil, diltiazem, amiodarone, ingrédients du pamplemousse, en particulier dans le jus de pamplemousse, etc.) En revanche, le CYP3A5, qui dégrade essentiellement les mêmes substances médicamenteuses que le CYP3A4, n’est presque jamais présent chez les Européens en raison d’une variante génétique, alors qu’il est généralement fonctionnel chez les Africains. Cependant, à l’exception de la loratadine, de l’azélastine et probablement de la cétirizine, le CYP3A4/5 ne joue aucun rôle dans les antihistaminiques.
Il était d’autant plus surprenant qu’une étude sur la fexofénadine ait mis en évidence un effet du jus de pamplemousse : Lorsque la fexofénadine était prise avec du jus de pamplemousse, les taux de fexofénadine diminuaient, en particulier peu après la prise, par rapport à la prise avec de l’eau (on aurait pu s’attendre à une augmentation en cas d’inhibition du CYP3A4 par les furanocoumarines du jus de pamplemousse). Cet effet peut s’expliquer par le fait que d’autres substances présentes dans le jus de pamplemousse (le flavonoïde naringine) inhibent le transporteur intestinal vers l’intérieur OATP1A2, nécessaire à l’absorption de la fexofénadine à partir de la lumière intestinale.
Une autre interaction, qui n’a pas encore été entièrement élucidée, concerne également la fexofénadine : l’administration concomitante d’itraconazole a entraîné des taux de fexofénadine beaucoup plus élevés . La fexofénadine n’étant pas métabolisée par le CYP3A, qui est puissamment inhibé par l’itraconazole, l’interaction a été expliquée par une inhibition de la glycoprotéine P, le transporteur qui jouerait un rôle important dans l’élimination de la fexofénadine. Il reste à déterminer si cette hypothèse est correcte et si d’autres antifongiques azolés entraînent également des augmentations de taux.
D’autre part, il convient de noter que les substances telles que la (lévo-)cétirizine sont principalement éliminées par voie rénale, de sorte que les restrictions de la fonction rénale doivent également conduire à des réductions de dose. Pour la lévocétirizine, par exemple, il est possible de réduire la dose dès une Pour les patients dont le taux de filtration glomérulaire est inférieur à 50 mL/min, l’administration de 5 mg tous les deux jours est prescrite ; en cas d’altération plus importante de la fonction rénale, l’intervalle entre les doses doit être encore plus long.
En Suisse, les services de pharmacologie clinique des hôpitaux universitaires peuvent fournir aux patients des informations sur les effets secondaires et les interactions, les adaptations de doses et d’autres problèmes liés aux médicaments.
Effets indésirables des antihistaminiques : Focus temps de QTc
Alors que la plupart des effets indésirables des antihistaminiques H1 sont dus à leur action sur les récepteurs H1, la plupart d’entre eux sont dus à leur action sur les récepteurs H2. (fatigue, baisse des performances cognitives et psychomotrices, augmentation de l’appétit) ou (pour les substances plus anciennes) par des effets sur le récepteur m-acétylcholine (sécheresse buccale, rétention urinaire, tachycardie), sur le récepteur alpha-adrénergique (hypotension, vertiges, tachycardie réflexe) ou sur le récepteur sérotonine (par ex. Si les effets de certains antihistaminiques H1 peuvent s’expliquer par une augmentation de l’appétit, on sait moins que certains antihistaminiques H1 inhibent également les canaux ioniques cardiaques, en particulier le canal IKr, qui module la sortie rapide du potassium lors de la repolarisation et peut ainsi entraîner un allongement de la repolarisation (de l’intervalle QT sur l’ECG), voire des tachycardies ventriculaires en torsades de pointes.
Dans l’ensemble, le potentiel des antihistaminiques H1 à prolonger l’intervalle QTc sur l’ECG a été mal étudié. L’intérêt pour cet effet secondaire potentiellement mortel a été suscité par lorsque plusieurs épisodes de torsades de pointes ont été rapportés pour les premiers antihistaminiques H1 de deuxième génération, la terfénadine et l’astémizole. Dans la plupart des cas , des surdoses avaient été prises ou des interactions augmentant la concentration n’avaient pas été prises en compte. Ces deux substances ont été retirées du marché en 1990 en raison d’arythmies ventriculaires. Des cas isolés d’allongement de l’intervalle QTc et parfois de torsades de pointes ont été rapportés pour la loratadine, fexofénadine et la cétirizine.
Les facteurs de risque généraux d’allongement de l’intervalle QTc sont présentés dans le tableau 3 . Il semble donc conseillé, en particulier chez les patients à risque sous traitement antihistaminique régulier et à fortes doses, de réaliser un ECG et de contrôler le potassium et le magnésium dans le sérum.

CONCLUSION POUR LA PRATIQUE
- Les antihistaminiques H1 de deuxième génération sont généralement bien tolérés.
- Un allongement de l’intervalle QTc est possible, surtout en cas de surdosage et de facteurs de risque.
- Le jus de pamplemousse ne doit pas être consommé avec la fexofénadine, car il diminue les concentrations de fexofénadine. L’itraconazole augmente les concentrations de fexofénadine, ce qui peut entraîner des symptômes de surdosage.
- Pour les antihistaminiques éliminés principalement par voie rénale, comme la (lévo-)cétirizine, la dose doit être adaptée en cas d’insuffisance rénale.
PD Dr. med. Alexander Jetter
Littérature :
- Simons FE, Simons KJ : Histamine and H1-antihistamines : celebrating a century of progress. J Allergy Clin Immunol 2011 ; 128 : 1139-1150.
- Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG : Effet de l’itraconazole sur la pharmacocinétique et la pharmacodynamie de la fexofénadine en relation avec le polymorphisme génétique MDR1. Clin Pharmacol Ther 2005 ; 78 : 191-201.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M : Le jus de pamplemousse réduit la biodisponibilité orale de la fexofénadine mais pas de la desloratadine. Clin Pharmacokinet 2002 ; 41 : 311-318.
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De Clerck F : La prolongation du QTc induite par les médicaments sous-estime dangereusement le potentiel proarythmique : leçons tirées de la terfénadine. J Cardiovasc Pharmacol 2011 ; 57 : 589-597.
- Compalati E, Baena-Cagnani R, Penagos M, Badellino H, Braido F, Gómez RM, Canonica GW, Baena-Cagnani CE : Systematic review on the efficacy of fexofenadine in seasonal allergic rhinitis : a meta-analysis of randomized, double-blind, placebo-controlled clinical trials. Int Arch Allergy Immunol 2011 ; 156 : 1-15.
PRATIQUE DU MÉDECIN DE FAMILLE 2013 ; 8(3) : 14-17