Un traitement précoce et adéquat peut avoir un effet positif sur l’évolution de cette maladie multisystémique rare. Cet article donne un bref aperçu des connaissances actuelles. Entre autres, l’identification de nouveaux biomarqueurs pourrait à l’avenir servir de base à un traitement personnalisé.
La dermatomyosite juvénile (DMJ) est une maladie systémique à médiation immunitaire qui touche les enfants et se caractérise par une faiblesse musculaire et des éruptions cutanées typiques [9]. D’autres systèmes d’organes, comme le cœur et les poumons, peuvent également être touchés et restent la principale cause de décès de ces patients. C’est la myopathie inflammatoire idiopathique la plus fréquente chez l’enfant, avec une incidence de 2-4/million/an.
Le processus inflammatoire se caractérise par l’infiltration des tissus concernés par des cellules du système immunitaire, telles que les cellules T et les cellules dendritiques plasmacytoïdes, ainsi que par une signature interféron [1]. La plupart des symptômes sont dus à une vasculopathie, qui entraîne un dysfonctionnement et une perte des cellules endothéliales. Grâce à une immunosuppression efficace, généralement un traitement combiné de méthotrexate sous-cutané et de corticostéroïdes oraux, la mortalité a fortement diminué (actuellement à environ 1%) et environ 50% des patients ne présentent qu’une seule poussée de la maladie sous traitement avant d’atteindre la rémission [2]. Les 50% restants présentent une évolution chronique, avec des poussées fréquentes, une morbidité accrue et une qualité de vie réduite [2].

Sous-phénotypage par biomarqueurs
La MJC est une maladie très hétérogène avec des phénotypes différents, divers systèmes d’organes pouvant être touchés à des degrés divers. Les auto-anticorps ont fait l’objet de recherches intensives ces dernières années et ont permis de mieux caractériser les divers phénotypes ; les auto-anticorps ont donc une valeur pronostique [3]. Le suivi de l’activité de la maladie et des lésions organiques provoquées par l’inflammation est un élément important de la prise en charge de ces jeunes patients, car il détermine les conséquences à long terme de cette maladie [4]. Différents outils sont alors à la disposition du clinicien, comme l’histopathologie des tissus musculaires et cutanés, la microscopie capillaire (Fig. 1), les scores cliniques pour évaluer l’atteinte musculaire et cutanée [5], et maintenant différents biomarqueurs sanguins [6,7]). Les scores cliniques permettant d’évaluer l’activité de la maladie nécessitent la coopération du patient, ce qui représente parfois un défi important chez les jeunes enfants. De même, ces scores cliniques ne sont que d’une utilité limitée pour détecter une inflammation sous-jacente ou pour distinguer d’autres causes de faiblesse musculaire, comme la toxicité des stéroïdes.

Lignes directrices pour l’optimisation de la gestion de la maladie
En 2012, l’initiative européenne SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) a été lancée dans le but d’uniformiser la prise en charge diagnostique et thérapeutique des enfants et adolescents atteints de maladies rhumatismales à travers l’Europe. Pour les MJD, ces recommandations ont été publiées en 2017 [6].
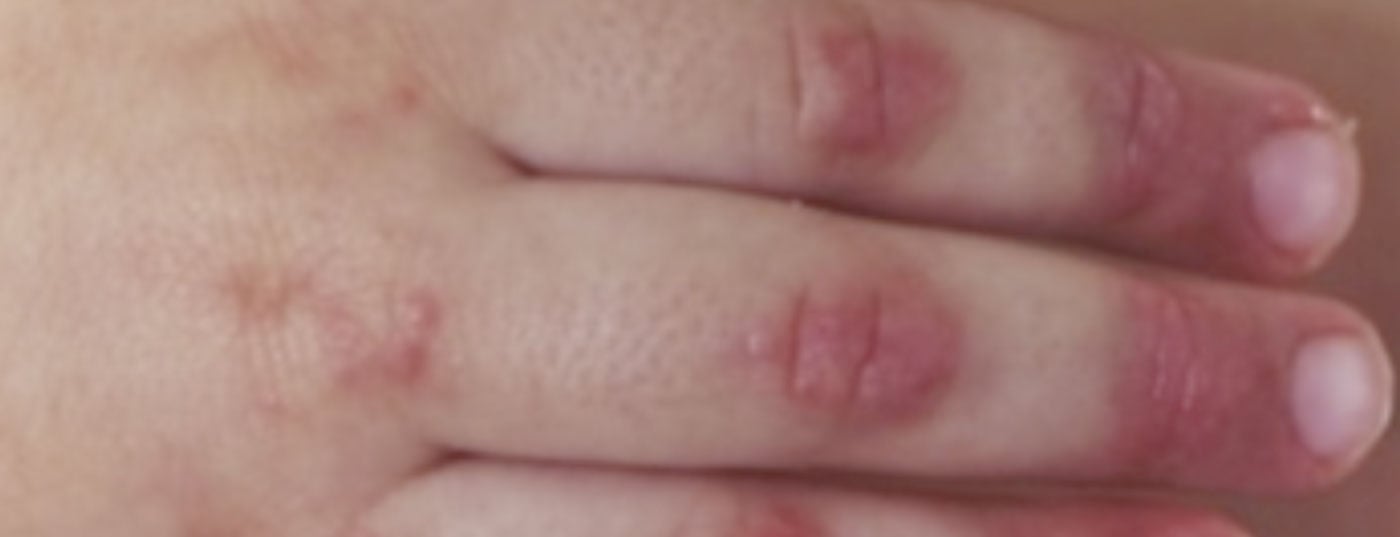
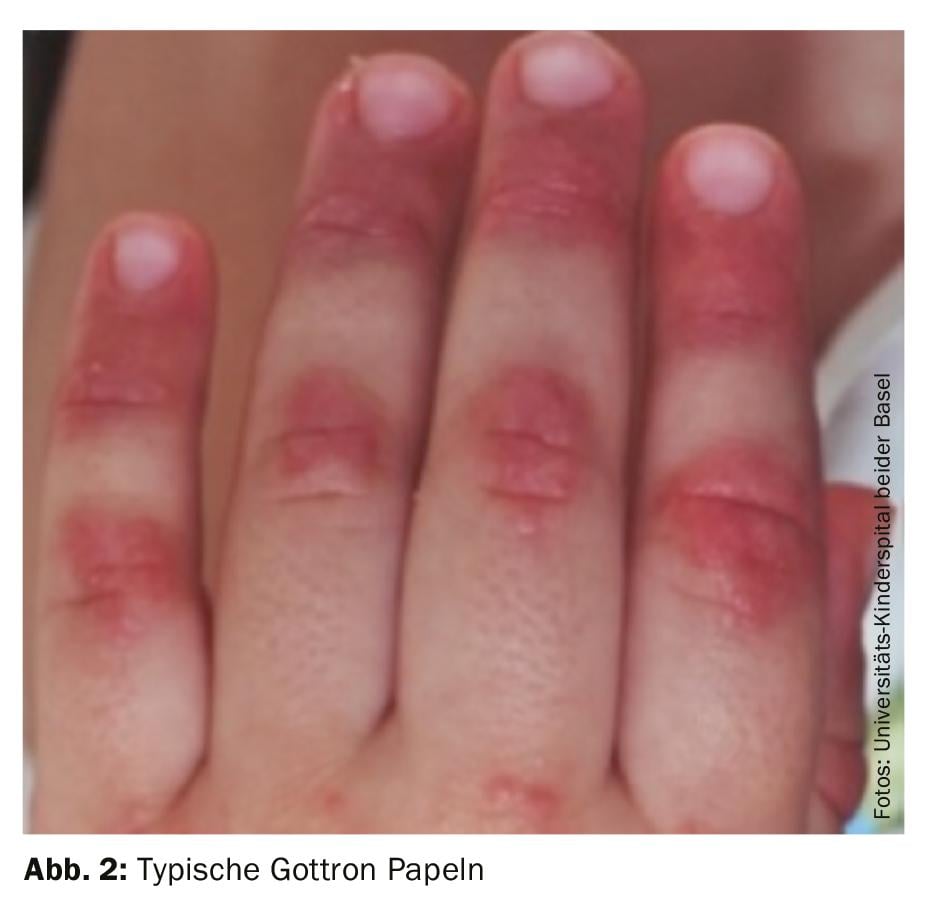
1. diagnostic : le diagnostic de dermatomyosite juvénile reste avant tout clinique avec des signes cutanés typiques (figure 2) et une faiblesse musculaire accentuée au niveau proximal. Par conséquent, en cas de suspicion de MJD, il convient d’orienter rapidement le patient vers un centre de compétence proche. Un traitement précoce et adéquat peut prévenir les dommages à long terme et augmenter les chances de rémission précoce. Les critères diagnostiques classiques de Bohan et Peter, publiés en 1975, comprennent cinq éléments :
- des manifestations cutanées caractéristiques
- faiblesse musculaire proximale
- biopsie musculaire typiquement pathologique
- augmentation des enzymes musculaires
- modifications myopathiques de l’électromyogramme (EMG)
→ Dans la pratique de la rhumatologie pédiatrique, l’EMG a été remplacé par l’IRM, qui n’est pas douloureuse (figure 3) [8].

De nouveaux critères de classification ont été publiés en 2017 ; ils renoncent par exemple à la biopsie musculaire en cas de manifestations cutanées pathognomoniques et incluent également les auto-anticorps (anti-Jo-1) dans les critères [9]. L’échographie est également de plus en plus utilisée, car elle ne nécessite pas de sédation chez les jeunes enfants.
La faiblesse musculaire doit être évaluée chez l’enfant à l’aide de différents instruments. Il existe des tests musculaires fonctionnels, appelés Children’s Myositis Activity Score (CMAS) [10], ainsi que des tests musculaires qui mesurent uniquement la force, appelés Manual Muscle Test (MMT) [11]. Ces deux tests sont utiles pour évaluer la faiblesse musculaire au moment du diagnostic ; ils servent également de paramètres d’évolution importants qui peuvent donner une indication significative de l’activité de la maladie. Les deux tests font partie des critères de rémission de 2013 [12].
Wienke et al. ont récemment validé plusieurs biomarqueurs (Galectine-9 et CXCL10) qui jouent un rôle non seulement dans le diagnostic mais aussi dans le suivi et qui pourraient éventuellement faciliter les décisions thérapeutiques au début de la maladie : Dans cette étude prospective, les patients présentant des taux élevés de ces biomarqueurs au moment du diagnostic ont présenté une évolution plus sévère au cours du suivi et pourraient donc potentiellement bénéficier d’un traitement plus intensif. En revanche, chez les patients n’ayant eu qu’une seule poussée, les valeurs s’étaient presque normalisées quelques mois seulement après le diagnostic sous traitement. En outre, il a été démontré que les marqueurs présentaient une nouvelle augmentation dans le sang avant même les résultats cliniques d’une poussée de la maladie [7].

2ème traitement : le traitement de première ligne reste une combinaison de stéroïdes et de mé-thotrexate, éventuellement de ciclosporine [6]. Le méthotrexate doit être maintenu pendant au moins 2 ans, les stéroïdes sont supprimés en cas d’évolution monophasique au cours de la 1ère année de traitement [6]. Si certains symptômes tels qu’une faiblesse musculaire importante (l’enfant ne peut plus se lever de son lit, dysphagie importante), une atteinte de la fonction myocardique ou pulmonaire ou des ulcères cutanés indiquent une évolution grave, il convient d’ajouter du cyclophosphamide. En l’absence d’amélioration, il convient d’ajouter des immunoglobulines intraveineuses ou de la ciclosporine A, ou encore de passer à des médicaments biologiques (rituximab, infliximab, adalimumab). D’un point de vue pédiatrique, des calcinoses croissantes du tissu sous-cutané ou musculaire (figures 3 et 4) sont le signe d’une inflammation systémique persistante qui justifie une intensification du traitement. Des greffes de cellules souches ont également été réalisées avec succès chez des patients atteints de MJC très sévère ; certains patients ont obtenu une rémission complète sans traitement. Lorsqu’une rémission clinique est obtenue, la calcinose peut disparaître complètement.
En cas de succès, ce traitement standard reste associé au risque d’effets secondaires indésirables, tels que des infections, une faiblesse musculaire induite par les stéroïdes ou des nausées hebdomadaires sous méthotrexate. A l’avenir, les nouveaux biomarqueurs mentionnés pourraient permettre une identification précoce d’une nouvelle poussée de la maladie ou une réduction plus rapide du traitement. Ces biomarqueurs pourraient ainsi contribuer à ouvrir la voie à un traitement personnalisé de cette maladie rare.
| Outil de diagnostic basé sur le web Entre-temps, un outil web est disponible qui, outre le diagnostic, donne également une probabilité de diagnostic avec la classification du sous-groupe. Ces critères ont été validés à la fois pour les enfants et les adultes en cas de suspicion de myopathie inflammatoire. http://www.imm.ki.se/biostatistics/calculators/iim/ |
Littérature :
- Piper CJM, et al : CD19(+)CD24(hi)CD38(hi) B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-alpha. Front Immunol 2018 ; 9 : 1372.
- Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD : Caractéristiques cliniques et évolution de la maladie chez les patients atteints de dermatomyosite juvénile. Int J Rheum Dis 2013 ; 16(5) : 561-567.
- Rider LG, Nistala K : The juvenile idiopathic inflammatory myopathies : pathogenesis, clinical and autoantibody phenotypes, and outcomes. J Intern Med 2016 ; 280(1) : 24-38.
- Huber A, Feldman BM : Résultats à long terme dans la dermatomyosite juvénile : comment en sommes-nous arrivés là et où allons-nous ? Curr Rheumatol Rep 2005 ; 7(6) : 441-446.
- Huber AM, et al : Validation préliminaire et signification clinique de l’outil d’évaluation cutanée dans la dermatomyosite juvénile. Arthritis Rheum 2008 ; 59(2) : 214-221.
- Enders FB, et al : Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017 ; 76(2) : 329-340.
- Wienke J, et al : Galectine-9 et CXCL10 comme biomarqueurs de l’activité de la maladie dans la dermatomyosite juvénile : une étude de cohorte longitudinale et une validation multicohorte. Arthritis Rheumatol 2019. doi.org/10.1002/art.40881
- Brown VE, et al. : An international consensus survey of the diagnostic criteria for juvenile dermatomyositis (JDM). Rheumatology (Oxford) 2006 ; 45(8) : 990-993.
- Lundberg IE, et al : 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017 ; 76(12) : 1955-1964.
- Huber AM, et al : Validation and clinical significance of the Childhood Myositis Assessment Scale for assessment of muscle function in the juvenile idiopathic inflammatory myopathies. Arthritis Rheum 2004 ; 50(5) : 1595-1603.
- Jain M, et al. : Fiabilité intra-contrôle et inter-contrôle du test musculaire manuel (MMT) en 10 points de la force chez les enfants atteints de myopathies inflammatoires idiopathiques juvéniles (MIIJ). Phys Occup Ther Pediatr 2006 ; 26(3) : 5-17.
- Lazarevic D, et al : The PRINTO criteria for clinically inactive disease in juvenile dermatomyositis. Ann Rheum Dis 2013 ; 72(5) : 686-693.
- Mathiesen PR, et al. : Aérobic fitness after JDM–a long-term follow-up study. Rheumatology (Oxford) 2013 ; 52(2) : 287-295.
DERMATOLOGIE PRATIQUE 2019 ; 29(4) : 26-29