Les cellules T dotées d’une spécificité tumorale grâce à l’expression d’un récepteur antigénique chimérique (chimeric antigen receptor ; CAR) prennent de plus en plus d’importance. Elles sont déjà de plus en plus utilisées dans la thérapie cellulaire adoptive pour lutter contre le cancer. Le grand avantage du transfert d’un CAR par rapport au transfert d’un récepteur de cellules T normales (T-cell receptor ; TCR) est qu’un CAR peut reconnaître la tumeur indépendamment du CMH.
Les cellules T dotées d’une spécificité tumorale grâce à l’expression d’un récepteur antigénique chimérique (chimeric antigen receptor ; CAR) prennent de plus en plus d’importance. Elles sont déjà de plus en plus utilisées dans la thérapie cellulaire adoptive pour lutter contre le cancer. Le concept CAR a été initialement développé à la fin des années 1980 par Zelig Eshhar (Weizmann Institute of Science, Rehovot, Israël) [1,2]. La plupart des CAR sont constitués d’une construction scFv dérivée d’anticorps qui se lie à l’antigène tumoral. (fragment variable à chaîne unique, qui est une protéine de fusion artificielle composée d’une partie variable d’une chaîne légère et d’une chaîne lourde d’une immunoglobuline) et la partie intracellulaire de la chaîne CD3ζ, qui est en do dièse est associé à un ou plusieurs domaines costimulatoires [3]. Cette structure modulaire permet l’activation des cellules T spécifiques de l’antigène en réponse à la reconnaissance spécifique des antigènes à la surface des cellules malignes, initiée par la liaison du scFv, et la signalisation subséquente via la chaîne CD3ζ ainsi que via le domaine costimulateur [3]. La costimulation se fait généralement soit par CD28 (superfamille des immunoglobulines), soit par 4-1BB (superfamille des récepteurs du TNF) [3]. Cependant, il existe de nombreux autres formats. Le grand avantage du transfert d’un CAR par rapport au transfert d’un récepteur de cellules T normales (T-cell receptor ; TCR) est qu’un CAR peut reconnaître la tumeur indépendamment du CMH.
Cette technologie a permis jusqu’à présent de développer des CARs ciblant différents antigènes de surface cellulaire sur des tumeurs solides ou hématologiques. Les cellules CAR-T, spécifiques d’antigènes cibles tels que CD19 sur les cellules B ou l’antigène de maturation des cellules B (BCMA) sur les plasmocytes, ont entraîné des régressions cliniques impressionnantes dans les leucémies, les lymphomes ou les myélomes dans plusieurs études cliniques [4–6]. Des résultats comme ceux-ci ont notamment conduit à à l’approbation de Tisagenlecleucel pour le traitement de la leucémie aiguë lymphoblastique à cellules B (LAL), d’Axicabtagen-Ciloleucel pour le traitement du lymphome agressif à cellules B non hodgkinien, Brexucabtagene-Autoleucel pour le traitement du lymphome à cellules du manteau, Lisocabtagene-Maraleucel contre le lymphome à grandes cellules B et Idecabtagene-Vicleucel et Ciltacabtagene-Autoleucel pour le traitement du myélome multiple par Administration américaine des denrées alimentaires et des médicaments (FDA) et la Agence européenne des médicaments (EMA) [3].
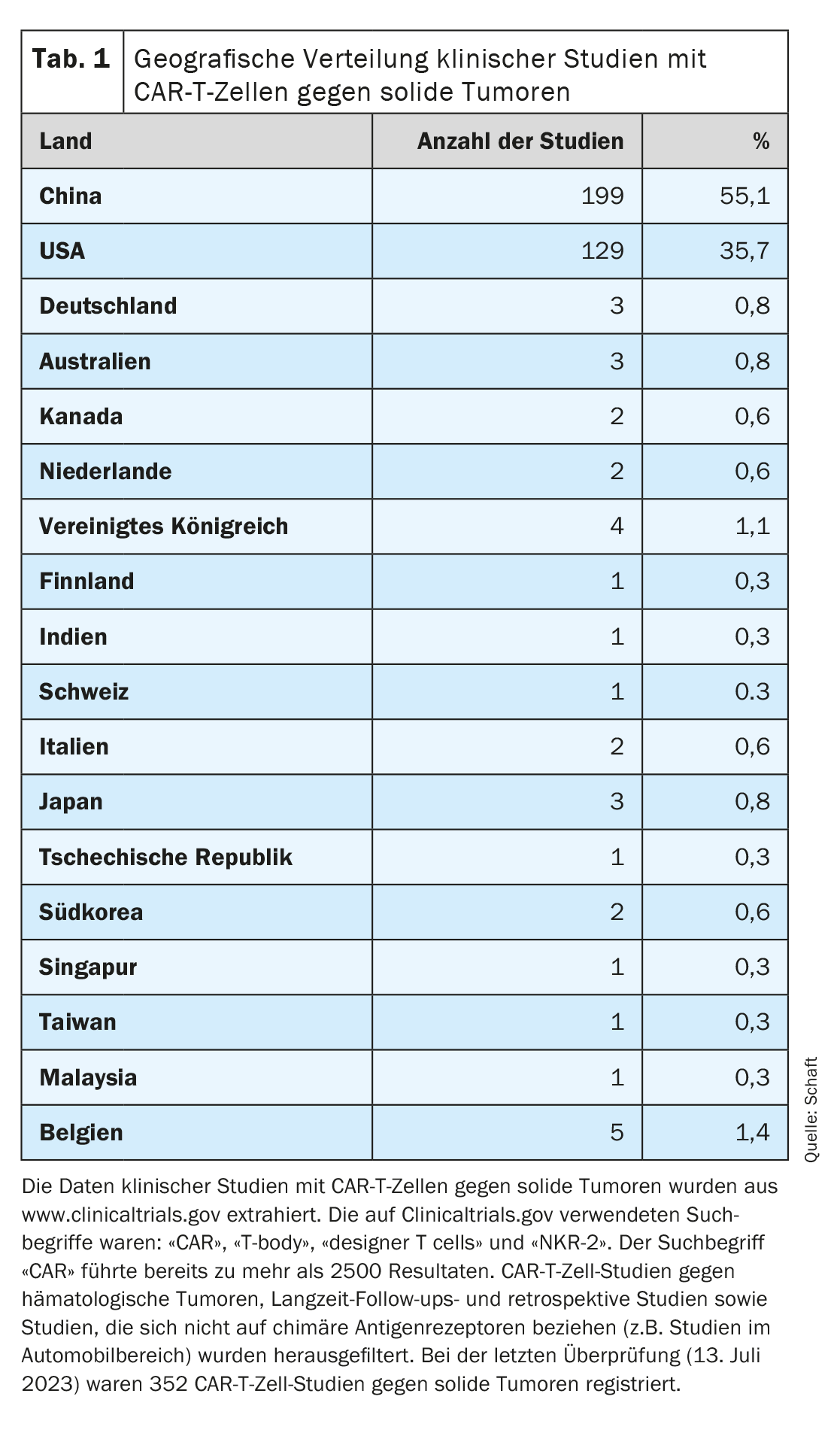
Comme la plupart des essais cliniques se concentrent sur l’élimination des tumeurs hématologiques, le développement des cellules CAR-T contre les tumeurs solides est à la traîne (revue détaillée sur [7–11]). Si l’on considère la répartition géographique des essais cliniques sur les cellules CAR-T contre les tumeurs solides enregistrés sur Clinicaltrials.gov (n=352 ; dernière évaluation le 13 juillet 2023), il apparaît clairement que la plupart de ces essais sont menés en Chine (n=199 ; 55,1%). En deuxième position, on trouve les États-Unis (n=129 ; 35,7%). Très peu d’études sont menées en Europe (Allemagne n=3, Suisse n=1), en Australie et dans le reste de l’Asie (ensemble n=33 ; 9,2%) (tableau 1).
Formats CAR
Depuis la publication du premier concept CAR par Zelig Eshhar en 1989 [1,2], les CAR n’ont cessé d’évoluer. Plusieurs générations de CAR ont ainsi vu le jour, basées sur la structure de base du concept CAR initial. Le CAR classique comprend toujours un scFv à base d’anticorps qui peut se lier à l’antigène tumoral. Dans les CAR de première génération (figure 1), le scFv est lié soit au domaine de signalisation intracellulaire de FcεRIγ, soit à CD3ζ, via un lieur flexible et un domaine transmembranaire [11,12].
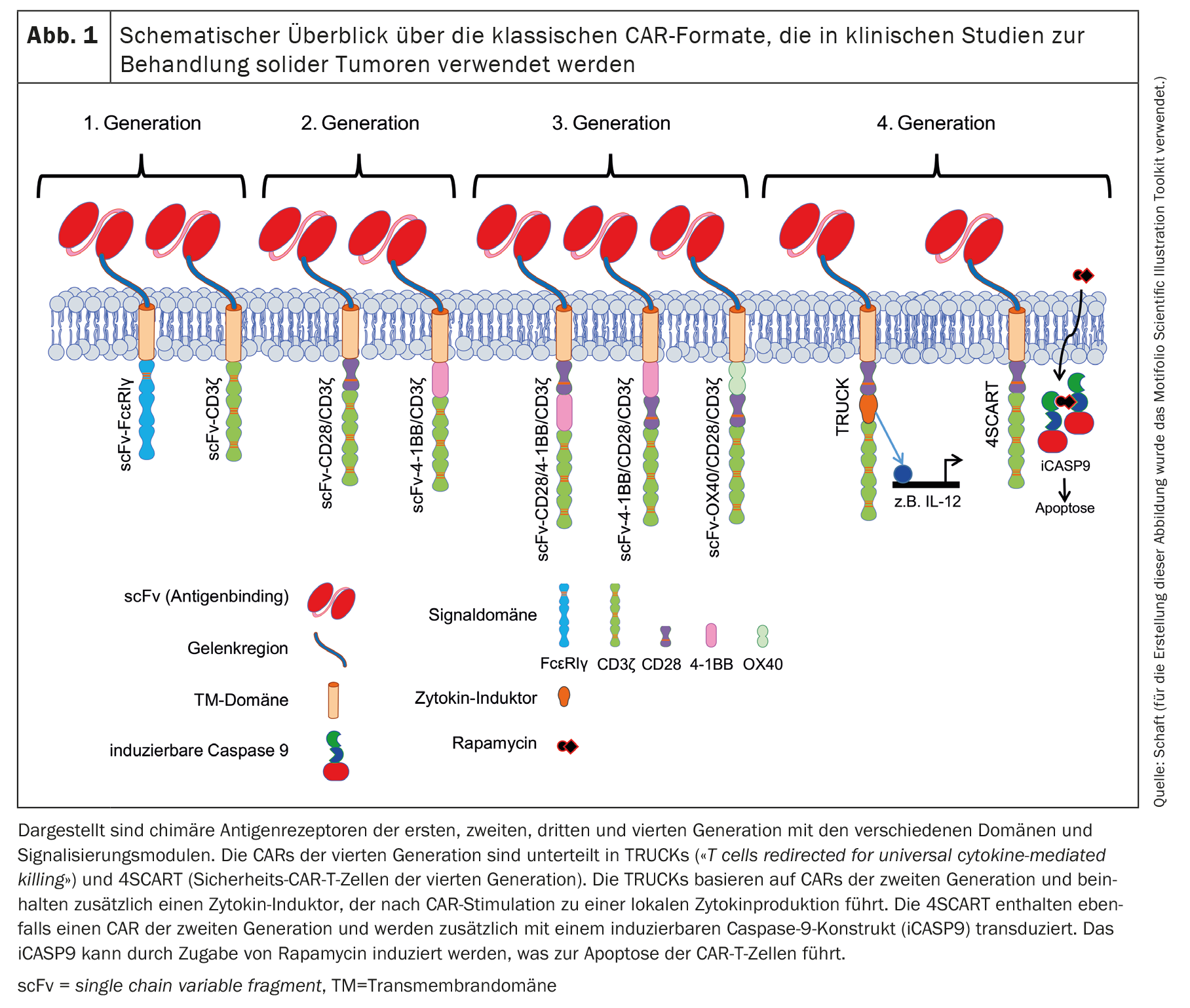
La plupart des études cliniques enregistrées sur les cellules CAR-T contre les tumeurs solides utilisent un CAR de deuxième génération [11,12], qui contient également un domaine de costimulation (Fig. 1). La costimulation est généralement assurée par CD28 ou 4-1BB [3]. La costimulation du CD28 soutient physiologiquement la production d’IL-2, -6, -10 et d’autres interleukines, ainsi que la progression du cycle cellulaire, la survie, la différenciation et la fonction cytolytique des lymphocytes T [13]. Dans de nombreuses études utilisant des CAR avec un domaine de signalisation CD28, des effets antitumoraux efficaces et rapides ont été observés. Cependant, ceux-ci étaient de courte durée et associés à une survie limitée in vivo par rapport, par exemple, aux CARs avec un domaine de signalisation 4-1BB [14]. La signalisation physiologique 4-1BB dans les cellules T augmente la progression et la prolifération du cycle cellulaire, la sécrétion de cytokines, le potentiel cytolytique des cellules T et inhibe la délétion clonale ainsi que la mort cellulaire induite par l’activation (activation-induced cell death, AICD) [15,16]. Les CARs contenant 4-1BB comme domaine de signalisation ont non seulement permis une activation cellulaire plus robuste, une persistance accrue in vivo, mais ont également favorisé la différenciation des cellules CAR-T vers des cellules à mémoire centrale(central memory cells) [4,14,17–24].
Les CARs de troisième génération [11,12] contiennent des combinaisons de domaines costimulants : CD28/4-1BB, 4-1BB/CD28 ou OX40/CD28 (Fig. 1) [25,26]. Les CAR de quatrième génération sont en principe des CAR de deuxième génération avec des caractéristiques supplémentaires. Les TRUCK (T cells redirected for universal cytokine-mediated killing) sont modifiées pour produire des cytokines de manière très localisée [27]. Les effets induits dépendent du type de cytokines sécrétées : Par exemple, l’IL-12 peut activer une réponse immunitaire innée contre la tumeur [28], provoque une moindre susceptibilité aux effets inhibiteurs des cellules T régulatrices (Tregs) [29] et augmente la sécrétion de cytokines et la prolifération des cellules T [30,31]. En revanche, l’IL-15 augmente l’activité antitumorale des cellules CAR-T [32].
Les cellules 4SCART (cellules T CAR de sécurité) constituent une autre variante de la quatrième génération. Ces cellules T sont transduites simultanément avec un CAR et une caspase 9 inductible (iCASP9), par mesure de précaution contre les événements indésirables. L’iCASP9 peut être induit par l’ajout de rapamycine, ce qui entraîne l’apoptose des cellules CAR-T. Les cellules CAR-T sont alors détruites.
Technologies de transfert
Une nécessité essentielle dans la production de cellules CAR-T est de trouver une méthode appropriée pour transférer le CAR dans les cellules T. Les cellules CAR-T peuvent être produites à l’aide d’une méthode de transfert de cellules CAR. Différentes méthodes existantes peuvent être utilisées à cet effet. La plupart des études cliniques utilisent une méthode de transfert viral (rétrovirale ou lentivirale) pour introduire le CAR de manière stable dans les cellules T. Les cellules T sont ensuite transférées dans le système immunitaire. Au cours de ce processus, un gène codant pour CAR est transporté du virus vers la cellule T, où il est intégré de manière stable dans l’ADN génomique. Les descendants de ces cellules transduites portent tous le gène CAR et peuvent exprimer le récepteur à la surface de leurs cellules. Les inconvénients de la transduction virale sont l’intégration arbitraire dans le génome de la cellule hôte, qui peut entraîner la destruction ou l’activation de certains gènes (c’est-à-dire la mutagenèse par insertion), et l’introduction de matériel/gènes viraux. Cette méthode peut entraîner des effets secondaires graves chez les patients traités avec des CAR-T cells. Lamers et al. ont par exemple décrit le développement de réponses immunitaires au transgène codant pour le récepteur et au vecteur rétroviral [33].
Certains essais cliniques utilisent un système d’administration de gènes non viral ou une méthode de transfert qui intègre le gène CAR à un site spécifique (par exemple, le système Sleeping Beauty Transposon [34–37], le système PiggyBac Transposon [36,37], CRISPR-Cas9 [38]). La transfection d’ADN ou d’ARN sont d’autres systèmes de transfert [39], mais ils ne conduisent pas à l’intégration de la séquence codante CAR dans le génome de la cellule hôte. L’expression transitoire du CAR qui en résulte présente certains avantages.
CAR-T cells against solid tumors – selection of antigen and safety precautions
L’utilisation clinique des CAR-T cells est à la traîne dans le traitement des tumeurs solides, comme décrit ci-dessus, contrairement au succès des CAR-T cells dans le traitement des tumeurs hématologiques. L’une des raisons est que CD19 et BCMA sont des antigènes cibles exprimés spécifiquement par les cellules B ou les plasmocytes, respectivement, et que leur élimination complète est relativement inoffensive. D’autres antigènes, en particulier sur les tumeurs solides, sont souvent exprimés sur les tissus sains, ce qui rend difficile le choix d’un antigène cible approprié.
Sélection de l’antigène
Les antigènes cibles idéaux sur les tumeurs solides combinent trois caractéristiques essentielles :
- une expression uniforme à la surface des cellules malignes, ce qui réduit le risque de variants d’échappement antigène-négatif.
- Une absence d’expression sur les cellules non malignes (c.-à-d. une expression exclusive sur les cellules tumorales), ce qui évite le risque d’effets secondaires graves et potentiellement mortels dus à l’activité on-target/off-tumor des CAR-T cells [40,41].
- Un rôle crucial en tant que pilote oncogène dans les cellules cancéreuses, empêchant la régulation négative de l’antigène en raison d’un avantage sélectif de survie des cellules malignes.
- De plus, la coexpression de l’antigène sur des cellules voisines au sein du microenvironnement tumoral (par exemple sur les vaisseaux associés à la tumeur, les fibroblastes et les macrophages) constitue une autre caractéristique positive d’un antigène cible idéal, car la structure d’approvisionnement de la tumeur peut être attaquée par le traitement spécifique à l’antigène.
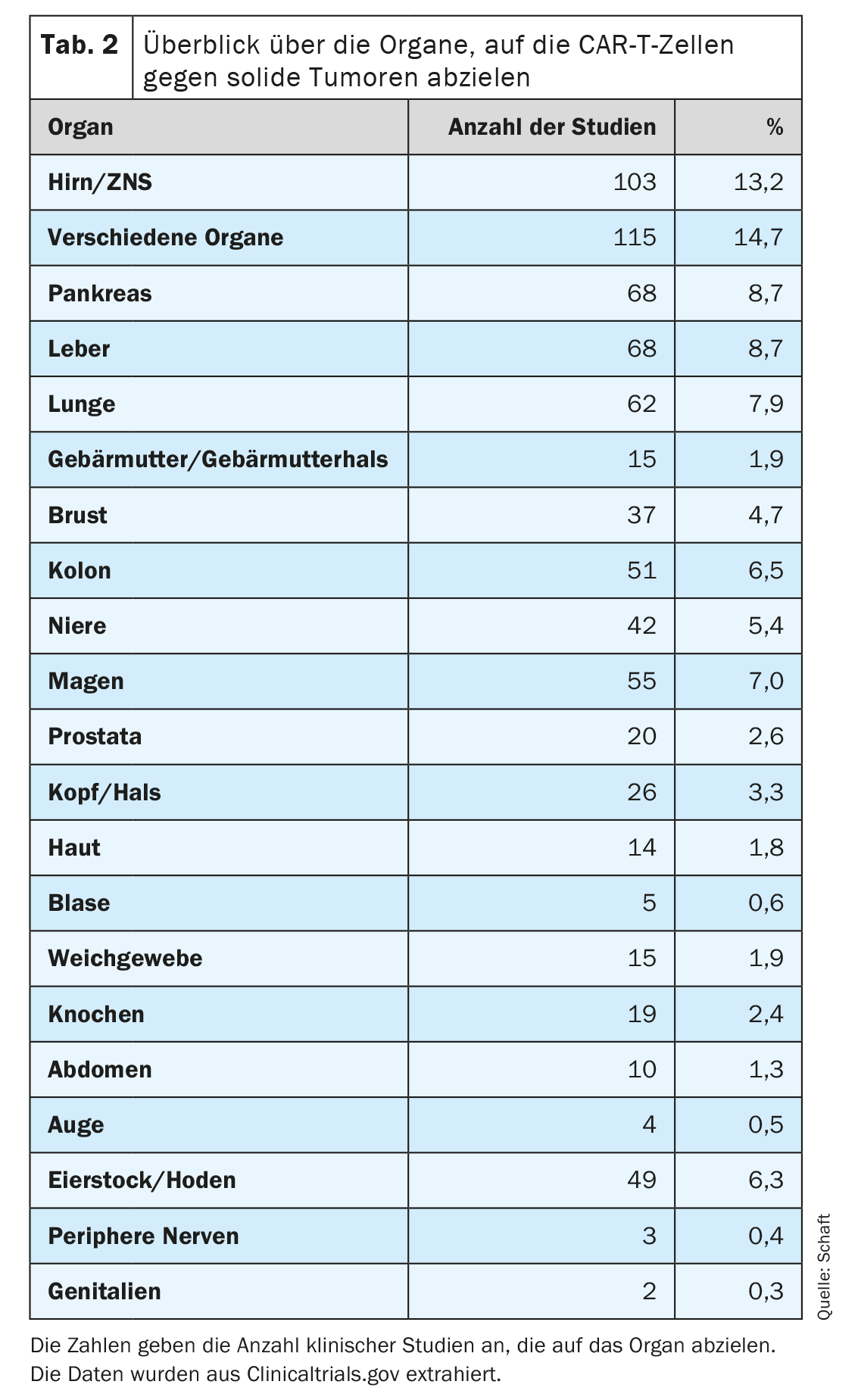
Le deuxième point en particulier constitue le plus grand problème pour le développement de cellules CAR-T contre les tumeurs solides, car la plupart des antigènes exprimés sur les tumeurs solides sont également exprimés sur des tissus sains importants. Cela peut entraîner une réaction indésirable on-target/off-tumor et des effets secondaires associés. Néanmoins, de nombreux types de tumeurs solides différentes (81 entités cancéreuses au total) dans 20 organes au total sont combattues avec des cellules CAR-T spécifiques de 63 antigènes différents (tableau 2). De nombreuses études cliniques portent en particulier sur des tumeurs du cerveau/SNC, du foie, du pancréas et des poumons (n=103, 68, 68 et 62, respectivement) ; Tab. 2). Cela pourrait être dû à l’importance des besoins médicaux et/ou à l’absence de thérapies alternatives efficaces pour les tumeurs dans les organes concernés.
Mesures de sécurité
Lorsqu’un antigène cible est reconnu par des cellules CAR-T transférées sur des tissus sains, des effets secondaires indésirables et graves peuvent survenir. Plusieurs stratégies ont été développées afin de pouvoir désactiver les cellules CAR-T le plus rapidement possible en cas de toxicité chez le patient. La rapamycine, une molécule capable d’induire la dimérisation de constructions, peut par exemple être utilisée pour activer une caspase 9 inductible. Dans 4SCART, ces constructions inductibles sont transférées dans les cellules T en même temps que le CAR, sous forme de ce que l’on appelle un suicide switch (figure 1). Après la dimérisation induite par la rapamycine, la caspase 9 initie l’apoptose des cellules T CAR. Cela permet également d’éliminer les activités indésirables/inattendues des cellules T contre les tissus sains(effets on-target/off-tumor) [42,43]. D’autres commutateurs possibles, tels que la stratégie herpès simplex virus thymidine kinase/ganciclovir (HSV-tk/GCV) [44,45] sont déjà utilisés [11].
Une mesure de sécurité particulière pour contourner une auto-immunité prolongée induite par une réaction On-Target/Off-Tumor du CAR est la transfection du CAR par électroporation de l’ARNm [11]. Nous avons déjà montré dans plusieurs publications que la transfection transitoire de cellules T avec des CARs par électroporation d’ARNm peut être un outil efficace et sûr dans l’immunothérapie du cancer [46-50]. Le procédé d’électroporation repose sur des mécanismes physico-chimiques complexes qui, par l’application de champs électriques, conduisent à la perforation de la membrane plasmique et permettent ensuite l’entrée de l’ARNm dans le cytosol [51]. L’utilisation de cellules CAR-T transfectées par ARN présente l’avantage de limiter l’expression du récepteur dans le temps, ce qui limite également la toxicité potentielle hors cible et sur cible/hors tumeur. La stratégie de transfert CAR-RNA est particulièrement attrayante dans les essais cliniques de phase 0/1 qui étudient de nouveaux antigènes tumoraux pour la thérapie CAR-T-cellulaire avec un profil de sécurité clinique inconnu.
Cellules CAR-T testées en clinique contre le mélanome choroïdien
Étonnamment, seules quatre études cliniques sur les CAR-T cells contre des tumeurs solides se concentrent sur l’entité œil (tableau 2). Parmi ces études, deux visent le rétinoblastome et deux le mélanome choroïdien. Le mélanome de la choroïde (uvéamélanome) est le type de cancer de l’œil le plus fréquent, et il se métastase chez jusqu’à 50% des patients. Les métastases se produisent principalement dans le foie et sont associées à une mauvaise survie moyenne d’environ 12 mois. Malgré d’énormes progrès dans le traitement du mélanome cutané métastatique par un blocage du point de contrôle immunitaire(ICB), il ne montre aucune efficacité dans le mélanome choroïdien. Seul l’agresseur bispécifique des cellules T Tebentafusp (un scFv spécifique de CD3 lié à un TCR soluble qui reconnaît un peptide gp100 présenté par HLA-A2), récemment approuvé, peut atténuer la progression et prolonger la survie globale dans un sous-groupe de patients atteints de mélanome choroïdien métastatique. Les effets positifs observés du tébentafusp sont de courte durée, avec une durée moyenne de survie globale de 22 mois et un taux de survie à trois ans de 24%. De plus, en raison de la restriction HLA-A2, seuls 50% des patients métastatiques sont éligibles à cette option de traitement. C’est pourquoi il existe également un besoin médical important pour des approches de traitement alternatives pour cette entité tumorale.
Actuellement, des patients atteints de mélanome choroïdien sont recrutés dans deux études cliniques sur les cellules CAR-T répertoriées dans la bibliothèque nationale internationale américaine de médecine (www.clinicaltrials.gov) (NCT03893019 contre Cluster of Differentiation 20 [CD20] et NCT03635632 contre Disialoganglioside GD2 [GD2]). Les deux études utilisent des cellules CAR-T qui ciblent des antigènes non spécifiques du mélanome.
La première étude clinique (phase 1 ; NCT03893019), qui utilise des cellules CAR-T spécifiques CD20 de deuxième génération (figure 2A), est sponsorisée par Miltenyi Biomedicine GmbH (Principal Investigator [PI]: Peter Borchmann ; Universitätsklinikum Köln) et recrute principalement des patients atteints de mélanome cutané. En outre, certains patients atteints de mélanome choroïdien sont également traités. CD20 est un antigène cible exprimé sur les cellules B normales et est typiquement utilisé comme antigène cible dans le lymphome non hodgkinien à cellules B [52]. Cependant, il est également exprimé sur un petit sous-groupe de cellules de mélanome [53,54]. Cependant, le ciblage d’un antigène exprimé uniquement sur un petit sous-groupe de cellules cancéreuses pourrait permettre à la tumeur d’échapper facilement au traitement par cellules CAR-T. Les cellules cancéreuses ne peuvent donc pas être traitées par le traitement par cellules CAR-T. Malheureusement, l’état actuel de cette étude clinique n’est pas connu.
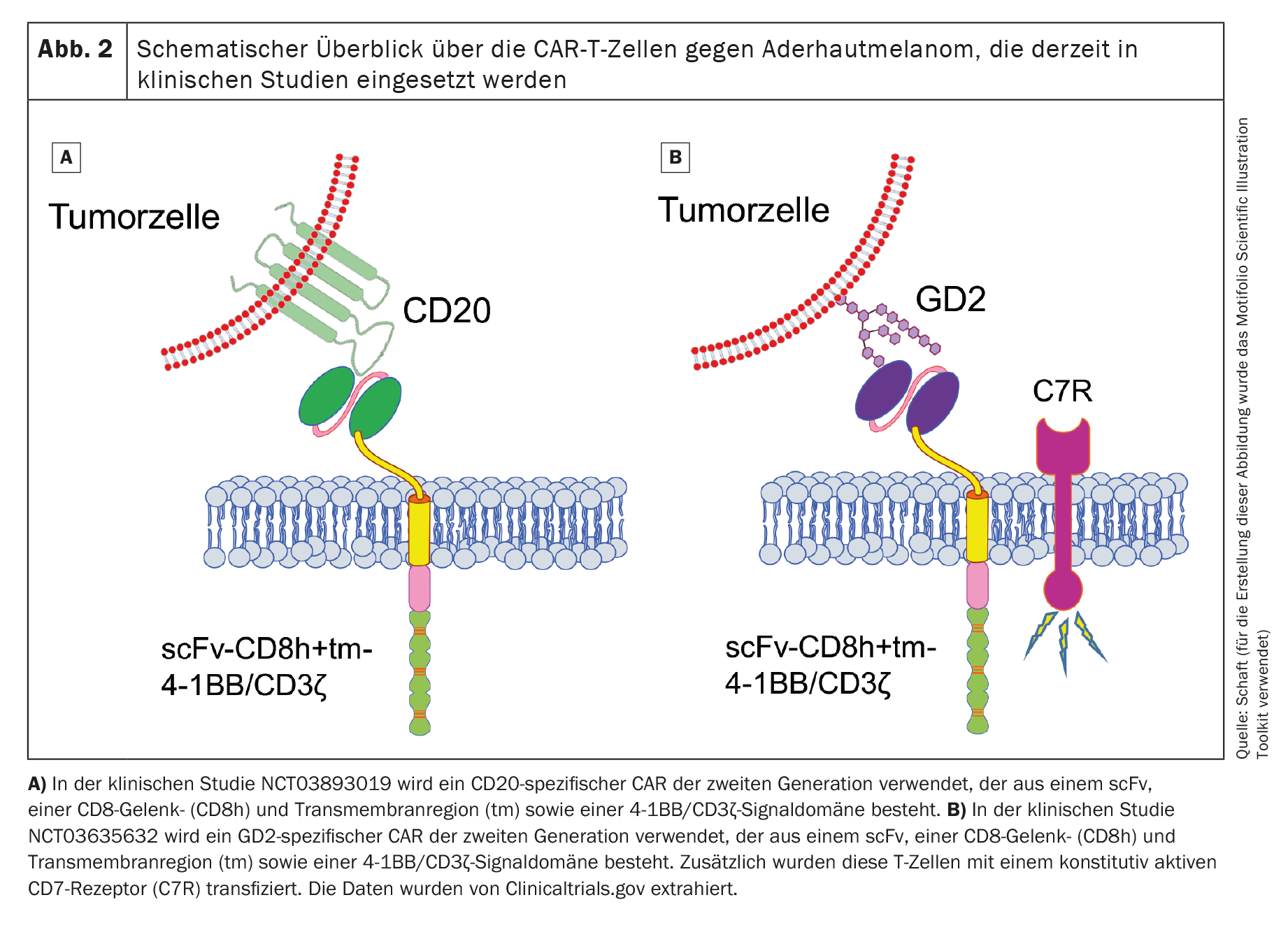
Le deuxième essai clinique (également de phase 1 ; NCT03635632) utilise des cellules CAR-T spécifiques de GD2 (figure 2B) et recrute des patients atteints de neuroblastome, de sarcome, de mélanome choroïdien, de cancer du sein ou d’autres types de cancer exprimant GD2. Cette étude, sponsorisée par le Baylor College of Medicine (PI : Bilal Omer ; Baylor College of Medicine), est actuellement active, mais aucun patient n’est actuellement recruté.
En plus du CAR de deuxième génération, les chercheurs transduisent également un récepteur de l’IL-7 constitutivement actif dans les cellules T afin de prolonger la survie des cellules CAR-T après le transfert adoptif. GD2 est exprimée, bien qu’en très faible quantité, dans le cervelet et les nerfs périphériques [55], ce qui rend le traitement avec des CAR-T cells spécifiques de GD2 très risqué si une réaction tumorale On-Target/Off est induite. Aucune donnée n’a non plus été publiée pour cette étude clinique.
En résumé, il existe un grand potentiel pour des essais cliniques avec des cellules CAR-T contre des antigènes spécifiques du mélanome (uvéal), pour lesquels le risque de réaction tumorale on-target/off est faible.
La recherche d’un meilleur antigène tumoral dans le mélanome choroïdien
Comme nous l’avons déjà mentionné dans la section “Les cellules CAR-T contre les tumeurs solides”, la prévention ou la réduction d’une éventuelle réaction on-target/off-tumor est une condition préalable à la recherche de nouveaux antigènes. Au niveau préclinique, l’accent est actuellement mis sur deux antigènes exprimés sur les mélanomes choroïdiens : le récepteur 2 du facteur de croissance épidermique humain (HER2) et le chondroïtine sulfate protéoglycane 4 (CSPG4).
HER2
HER2 est un membre de la famille ErbB des récepteurs tyrosine kinases (EGFR [ErbB-1], HER2 [/neu] [ErbB-2], Her 3 [ErbB-3] et Her 4 [ErbB-4]). Les mutations dans HER2 entraînent une surexpression qui conduit à une activation constitutive et à la division cellulaire incontrôlée qui en résulte. C’est particulièrement vrai pour le cancer du sein, mais aussi pour d’autres types de cancer, comme le cancer de l’ovaire ou les gliomes [56–58].
Comme nous l’avons déjà mentionné, l’utilisation de cellules CAR-T est une arme à double tranchant, car l’efficacité de ces cellules peut également se retourner contre le patient [59]. On ne peut jamais exclure qu’un type de cellule rare mais essentiel exprime l’antigène dans un tissu sain. Des chercheurs du National Cancer Institute ont rapporté un cas qui illustre le potentiel mortel de la toxicité tumorale On-Target/Off de l’antigène HER2. Peu après la perfusion de cellules CAR-T HER2-spécifiques, des symptômes cliniques de syndrome de détresse respiratoire aiguë ont été observés chez un patient atteint d’un cancer colorectal métastatique, nécessitant une respiration artificielle [60]. Malheureusement, le patient est décédé cinq jours après l’apparition de la détresse respiratoire aiguë [60]. La cause du décès était probablement le résultat d’une toxicité on-target/off-tumor causée par de faibles niveaux de HER2 sur les cellules épithéliales dans les poumons. De manière remarquable, le ROC était basé sur le trastuzumab, un anticorps monoclonal approuvé par la FDA, qui a été utilisé à grande échelle sans entraîner de toxicité pulmonaire grave [61]. Cela souligne la nécessité de sélectionner très soigneusement l’antigène cible pour une thérapie CAR-T-cell.
CSPG4
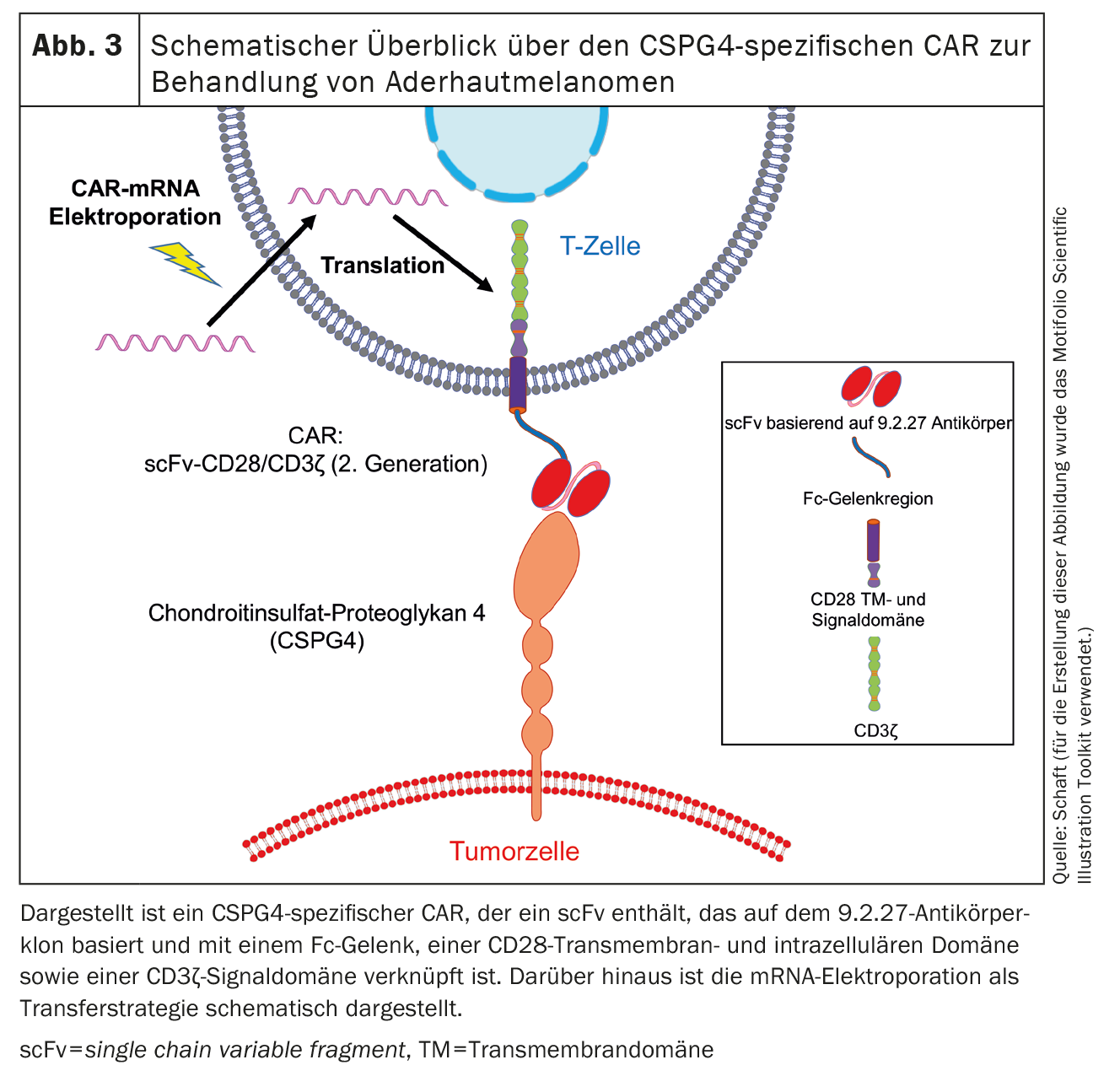
Le deuxième antigène exprimé sur les mélanomes choroïdiens est le chondroïtine sulfate protéoglycane 4 (CSPG4) (figure 3), anciennement connu sous le nom de chondroïtine sulfate protéoglycane associé au mélanome (MCSP) ou antigène associé au mélanome de haut poids moléculaire (HMW-MAA). CSPG4 est une protéine transmembranaire de type 1 à passage unique et a été découverte par Ralph Reisfeld [72]. Nous, ainsi que d’autres groupes de travail [47-50, 62-71], avons principalement travaillé sur le CSPG4. L’expression de CSPG4 est associée à une augmentation de la prolifération et de la survie des cellules tumorales. Ce processus est initié par l’activation de la voie de signalisation MAPK et la présentation croisée de facteurs de croissance [73]. En outre, la CSPG4 joue un rôle dans la motilité cellulaire et l’infiltration tissulaire en raison de son association avec le cytosquelette d’actine et de sa liaison à diverses intégrines et composants de la matrice extracellulaire [74]. En outre, CSPG4 est impliqué dans la formation du placenta [75], l’angiogenèse [76], la formation des réseaux neuronaux [77], le turn-over des kératinocytes et l’homéostasie des cellules souches épidermiques [78].
Plusieurs publications ont décrit l’expression de CSPG4 sur des tissus non pathologiques tels que les précurseurs des cellules du follicule pileux et de l’épiderme, les cellules endothéliales et les péricytes activés (mais pas sur les vaisseaux matures) [79,80], les chondrocytes du cartilage articulaire [81], les cellules musculaires lisses [82] et les cellules de la synapse neuromusculaire du muscle squelettique humain postnatal [83]. Cependant, CSPG4 est exprimé beaucoup plus faiblement sur les tissus sains que sur les cellules tumorales [62,73,84].
Beard et al. ont montré que le CSPG4 était détecté au niveau de l’ARN dans un grand nombre de tissus normaux : notamment le système nerveux central, l’œil, la peau, le tissu adipeux, les vaisseaux sanguins, la vessie, le tractus gastro-intestinal, l’utérus, la prostate, la rate et le thymus [85]. En moyenne, l’ARN CSPG4 est 6,6 fois plus surexprimé dans les tissus malins (mélanome) par rapport aux tissus sains [85]. Ces résultats ont confirmé les travaux antérieurs d’Erfurt et al. qui ont montré que, bien que l’ARNm du CSPG4 ait été détecté dans certains échantillons de tissus normaux, son expression était nettement plus élevée dans les échantillons de mélanome cutané et de mélanome choroïdien [86].
Les colorations immunohistochimiques et les puces à ADN en phase inverse ont montré qu’une expression concrète de CSPG4 au niveau protéique n’a été détectée que dans quelques échantillons de l’intestin grêle [63]. Aucune expression de la protéine CSPG4 n’a été détectée dans les tissus suivants : Cerveau, nerfs périphériques, peau, mésothélium, sein, cœur, rein, glandes surrénales, foie, poumons, ganglions lymphatiques, muscles, ovaires, pancréas, œsophage, prostate, rate, estomac, utérus et thyroïde [62,63]. En revanche, CSPG4 est exprimé sur presque toutes les cellules de mélanome cutané [87–90]. Les mélanomes choroïdiens [91,92] et certaines autres tumeurs comme les sarcomes, les astrocytomes, les gliomes, les neuroblastomes [93-96], les leucémies [97–101] et le cancer du sein triple négatif expriment également CSPG4 [102]. Dans beaucoup de ces pathologies malignes, l’expression de CSPG4 est associée à un mauvais pronostic et à une croissance agressive de la tumeur [103].
En outre, CSPG4 est considéré comme un antigène cible tumoral primaire [84], car il joue un rôle dans la métastase des mélanomes [104] et est exprimé sur les péricytes activés pendant l’angiogenèse dans les tumeurs et en cas d’hypoxie [105–107]. Ce dernier permet de cibler le système vasculaire tumoral. Plus important encore, CSPG4 agit comme un moteur oncogénique dans le mélanome, favorisant la croissance et la survie des cellules malignes après l’activation de différentes voies de signalisation [73]. Par conséquent, la tumeur ne peut pas simplement réguler à la baisse le CSPG4 pour échapper à un traitement ciblant le CSPG4.
C’est pourquoi plusieurs groupes ont déjà choisi CSPG4 comme antigène cible et ont introduit des CAR spécifiques à CSPG4 dans les cellules T par différents mécanismes. Les CAR spécifiques au CSPG4 de différents formats, transduits viralement dans les cellules T, ont entraîné une forte cytotoxicité des cellules T in vitro . Dans des modèles animaux, les cellules T transférées de manière adoptive ont réagi à différentes tumeurs exprimant le CSPG4, telles que le mélanome, le cancer du sein, le mésothéliome, le glioblastome et l’ostéosarcome [47–50,62–71]. Geldres et al. ont transduit de manière rétrovirale un CAR de deuxième génération spécifique au CSPG4 dans des cellules T. In vitro, ces cellules CAR T spécifiques du CSPG4 ont été capables de reconnaître et de lyser des cellules de mélanome de manière spécifique à l’antigène [65]. De plus, la liaison de l’antigène au CAR a entraîné une sécrétion prononcée d’IL-2 et d’IFNγ. In vivo , le transfert de cellules CAR-T spécifiques du CSPG4 chez des souris porteuses de cellules de mélanome a entraîné un ralentissement significatif de la croissance de la tumeur et une amélioration de la survie globale des souris [65]. La même publication décrit l’absence de réactivité positive des cellules CAR-T spécifiques du CSPG4 par rapport à des tissus normaux avec un ARN CSPG4 détectable, mais sans expression de la protéine CSPG4 [65]. Ils ont montré que les cellules CAR-T spécifiques au CSPG4 n’exerçaient pas de cytotoxicité significative sur les lignées de cellules épithéliales primaires de la prostate, des poumons et des reins [65]. Par conséquent, les analyses d’expression de Beard et al. [63,85], atténuent les préoccupations déjà décrites ci-dessus concernant latoxicité on-target/off-tumor induite par les cellules CAR-T spécifiques, car l’expression de CSPG4 au niveau protéique est nécessaire pour induire une réactivité indésirable des cellules CAR-T. En outre, l’expression de CSPG4 au niveau protéique est nécessaire pour induire une réactivité indésirable des cellules CAR-T.
Jusqu’à présent, nous avons utilisé la méthode de transfection d’ARNm par électroporation pour introduire des CAR dans les cellules T. Nous avons également utilisé la méthode de transfection d’ARNm par électrophorèse. Dans le cadre de ce projet, nous avons déjà testé plusieurs CARs spécifiques de CSPG4 et observé que les cellules CAR-T transfectées par ARNm sont capables d’éliminer les cellules tumorales de manière spécifique à l’antigène. La cinétique d’expression des CARs transférés par électroporation, dépend du squelette CAR [47]. Un CAR présentant à la fois une forte expression sur les cellules T et une forte réactivité anti-cellules tumorales a été identifié. Ce CAR contient un scFv basé sur le clone d’anticorps 9.2.27, lié à un espaceur Fc, un domaine transmembranaire et intracellulaire CD28 et un domaine de signalisation CD3ζ [47](Fig. 3). Des expériences in vivo sur des souris immunodéficientes à chaîne Rag-/-/ ordinaire γ-/- ont montré que les cellules CAR-T spécifiques du CSPG4 transfectées prolongeaient significativement la durée moyenne de survie des souris [47].
Afin de passer à l’application clinique des cellules CAR-T spécifiques au CSPG4, la production de cellules CAR-T à l’échelle clinique par transfection d’ARNm d’un CAR, dans le respect total des BPF, a été établie au laboratoire [50]. Cela a montré qu’il était possible de produire de manière répétée un nombre suffisant de cellules T transfectées par CAR et spécifiques au CSPG4 de haute pureté. Ces cellules T modifiées présentent une très grande efficacité de transfection, une expression CAR élevée et une grande efficacité dans l’élimination des cellules cibles du mélanome [50].
Bien que CSPG4 soit un antigène cible tumoral primaire, en particulier dans le mélanome cutané, nous avons également constaté qu’il n’était pas exprimé sur plusieurs lignées cellulaires de mélanome choroïdien que nous avons testées. Nous avons donc établi une plate-forme combinée in silico/in vitro pour identifier de nouveaux antigènes de surface cellulaire spécifiques des tumeurs du mélanome choroïdien. Grâce à cette plateforme, nous avons identifié une protéine candidate comme antigène cible approprié dans le mélanome choroïdien pour le développement d’autres CAR. Actuellement, les CARs générés, qui peuvent se lier à cette protéine candidate, sont testés pour leur fonctionnalité et leur spécificité.
Perspectives
L’utilisation des CAR-T cells dans le traitement des tumeurs solides offre de grandes opportunités. Des études précliniques et cliniques supplémentaires sont nécessaires pour répondre au besoin médical élevé de traiter des entités cancéreuses solides (comme le mélanome choroïdien).
Les futurs essais cliniques devraient se concentrer sur le test de nouveaux formats de CAR. Il s’agit notamment de tester de nouveaux domaines extracellulaires de liaison à l’antigène et de nouveaux domaines intracellulaires de signalisation [109], mais aussi de tester des formats qui améliorent la sécurité d’utilisation des cellules CAR-T [108]. En outre, de nouveaux véhicules cellulaires pour le transfert CAR [109,110] promettent d’élargir l’applicabilité. Par exemple, la possibilité d’utiliser par défaut des cellules CAR-NK [111] ou des cellules CAR-T allogéniques [112] peut réduire le coût de la thérapie par cellules CAR et rendre ainsi cette thérapie accessible à davantage de patients.
De plus, des antigènes plus spécifiques aux tumeurs devraient être trouvés afin d’éviter les réactions on-target/off-tumor. Les antigènes exprimés sur le stroma tumoral, où ils peuvent être attaqués par les cellules CAR-T, sont très prometteurs dans ce domaine [113]. Le ciblage de plusieurs antigènes par une cellule CAR-T (c’est-à-dire l’expression de différents CAR spécifiques de différents antigènes sur une seule cellule) peut augmenter la spécificité tumorale et réduire le risque d’effets hors cible. Il en va de même pour le modèle lorsque les modules de signalisation intracellulaire sont partagés entre les différents CAR afin d’améliorer le profil de sécurité des cellules CAR-T. Les CAR-T sont des cellules qui ont été conçues pour être utilisées dans le cadre d’une thérapie cellulaire. De plus, cela rend moins probable l’apparition de variants de perte d’antigènes des tumeurs.
En outre, des thérapies combinant des cellules CAR-T avec différentes petites molécules ou des anticorps monoclonaux pour éviter les mécanismes d’échappement tumoral et augmenter l’activité antitumorale sont déjà testées cliniquement pour de nombreuses tumeurs hématologiques (aperçu détaillé dans [114,115]). De telles combinaisons sont également prometteuses pour le traitement des tumeurs solides et doivent être testées dans des essais cliniques dans un avenir proche.
N.S. a effectué des recherches sur clinicaltrials.gov, a révisé les illustrations et a rédigé le manuscrit. S.H. a édité le manuscrit. N.S. et S.H. ont conçu ensemble les questions de formation continue CME.
Littérature :
- Gross G, Gorochov G, Waks T, Eshhar Z: Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc 1989; 21(1 Pt 1): 127–130.
- Gross G, Waks T, Eshhar Z: Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989; 86(24): 10024–10028.
- June CH, Sadelain M: Chimeric Antigen Receptor Therapy. N Engl J Med 2018; 379(1): 64–73.
- Maude SL, Laetsch TW, Buechner J, et al.: Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018; 378(5): 439-448.
- Neelapu SS, Locke FL, Go WY: CAR T-Cell Therapy in Large B-Cell Lymphoma. N Engl J Med 2018; 378(11): 1065.
- Schuster SJ, Bishop MR, Tam CS, et al.: Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med 2019; 380(1): 45–56.
- Wang Z, Guo Y, Han W: Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 2017; 8(12): 896–925.
- Han S, Latchoumanin O, Wu G, et al.: Recent clinical trials utilizing chimeric antigen receptor T cells therapies against solid tumors. Cancer Lett 2017; 390: 188–200.
- Yeku O, Li X, Brentjens RJ: Adoptive T-Cell Therapy for Solid Tumors. Am Soc Clin Oncol Educ Book 2017; 37: 193–204.
- Arabi F, Torabi-Rahvar M, Shariati A, et al.: Antigenic targets of CAR T Cell Therapy. A retrospective view on clinical trials. Exp Cell Res 2018; 369(1): 1–10.
- Schaft N: The Landscape of CAR-T Cell Clinical Trials against Solid Tumors-A Comprehensive Overview. Cancers (Basel) 2020; 12(9).
- Holzinger A, Abken H: CAR T Cells: A Snapshot on the Growing Options to Design a CAR. Hemasphere 2019; 3(1): e172.
- Boomer JS, Green JM: An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol 2010; 2(8): a002436.
- Kawalekar OU, O’Connor RS, Fraietta JA, et al.: Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016; 44(2): 380–390.
- Cannons JL, Choi Y, Watts TH: Role of TNF receptor-associated factor 2 and p38 mitogen-activated protein kinase activation during 4-1BB-dependent immune response. J Immunol 2000; 165(11): 6193–6204.
- Lee HW, Nam KO, Park SJ, Kwon BS: 4-1BB enhances CD8+ T cell expansion by regulating cell cycle progression through changes in expression of cyclins D and E and cyclin-dependent kinase inhibitor p27kip1. Eur J Immunol 2003; 33(8): 2133–2141.
- Zhao Z, Condomines M, van der Stegen SJC, et al.: Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015; 28(4): 415–428.
- Milone MC, Fish JD, Carpenito C, et al.: Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17(8): 1453–1464.
- Lim WA, June CH.: The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017; 168(4): 724-740.
- Roselli E, Frieling JS, Thorner K, et al.: CAR-T Engineering: Optimizing Signal Transduction and Effector Mechanisms. BioDrugs 2019; 33(6): 647–659.
- Hombach AA, Holzinger A, Abken H.: The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr Mol Med 2013; 13(7): 1079–1088.
- Sadelain M, Brentjens R, Riviere I: The basic principles of chimeric antigen receptor design. Cancer Discov 2013; 3(4): 388-398.
- Redeker A, Arens R: Improving Adoptive T Cell Therapy: The Particular Role of T Cell Costimulation, Cytokines, and Post-Transfer Vaccination. Front Immunol 2016; 7: 345.
- Weinkove R, George P, Dasyam N, McLellan AD.: Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology 2019; 8(5): e1049.
- Ceppi F, Rivers J, Annesley C, et al.: Lymphocyte apheresis for chimeric antigen receptor T-cell manufacturing in children and young adults with leukemia and neuroblastoma. Transfusion 2018; 58(6): 1414–1420.
- Li W, Guo L, Rathi P, et al.: Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum Gene Ther 2017; 28(5): 437–448.
- Chmielewski M, Hombach AA, Abken H: Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 2014; 257(1): 83–90.
- Chmielewski M, Kopecky C, Hombach AA, Abken H.: IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71(17): 5697–5706.
- Pegram HJ, Lee JC, Hayman EG, et al.: Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012; 119(18): 4133-4141.
- Koneru M, Purdon TJ, Spriggs D, et al.: IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015; 4(3): e994446.
- Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ.: A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med 2015; 13: 102.
- Xu A, Bhanumathy KK, Wu J, et al.: IL-15 signaling promotes adoptive effector T-cell survival and memory formation in irradiation-induced lymphopenia. Cell Biosci 2016; 6: 30.
- Lamers CH, Willemsen R, van Elzakker P, et al.: Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 2011; 117(1): 72–82.
- Magnani CF, Tettamanti S, Alberti G, et al.: Transposon-Based CAR T Cells in Acute Leukemias: Where are We Going? Cells 2020; 9(6).
- Hudecek M, Ivics Z: Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr Opin Genet Dev 2018; 52: 100–108.
- Tipanee J, VandenDriessche T, Chuah MK.: Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum Gene Ther 2017; 28(11): 1087–1104.
- Vargas JE, Chicaybam L, Stein RT, et al.: Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J Transl Med 2016; 14(1): 288.
- Ran FA, Hsu PD, Wright J, et al.: Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013; 8(11): 2281–2308.
- Birkholz K, Hombach A, Krug C, et al.: Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther 2009; 16(5): 596–604.
- Lamers CH, Sleijfer S, et al.: Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013; 21(4): 904–912.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843-851.
- Stavrou M, Philip B, Traynor-White C, et al.: A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol Ther 2018; 26(5): 1266–1276.
- Di Stasi A, Tey SK, Dotti G, et al.: Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365(18): 1673–1683.
- Moolten FL.: Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res 1986; 46(10): 5276–5281.
- Beltinger C, Fulda S, Kammertoens T, et al.: Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci U S A 1999; 96(15): 8699–8704.
- Harrer DC, Simon B, Fujii SI, et al.: RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: a safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17(1): 551.
- Krug C, Birkholz K, Paulus A, et al : La stabilité et l’activité des récepteurs d’antigènes chimériques (CARs) spécifiques au MCSP dépendent du domaine de liaison à l’antigène scFv et de la dorsale protéique. Cancer Immunol Immunother 2015 ; 64(12) : 1623-1635.
- Dörrie J, Babalija L, Hoyer S, et al.: BRAF and MEK Inhibitors Influence the Function of Reprogrammed T Cells: Consequences for Adoptive T-Cell Therapy. Int J Mol Sci 2018; 19(1).
- Harrer DC, Schuler G, Dörrie J, Schaft N : CSPG4-Specific CAR T Cells for High-Risk Childhood B Cell Precursor Leukemia. Int J Mol Sci 2019 ; 20(11).
- Wiesinger M, Marz J, Kummer M, et al.: Clinical-Scale Production of CAR-T Cells for the Treatment of Melanoma Patients by mRNA Transfection of a CSPG4-Specific CAR under Full GMP Compliance. Cancers (Basel) 2019; 11(8).
- Shi J, Ma Y, Zhu J, et al : A Review on Electroporation-Based Intracellular Delivery. Molecules 2018 ; 23(11).
- Ernst M, Oeser A, Besiroglu B, et al.: Chimeric antigen receptor (CAR) T-cell therapy for people with relapsed or refractory diffuse large B-cell lymphoma. Cochrane Database Syst Rev 2021; 9(9): Cd013365.
- Pinc A, Somasundaram R, Wagner C, et al.: Targeting CD20 in melanoma patients at high risk of disease recurrence. Mol Ther 2012; 20(5): 1056–1062.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474–2479.
- Brignole C, Marimpietri D, Pagnan G, et al.: Neuroblastoma targeting by c-myb-selective antisense oligonucleotides entrapped in anti-GD2 immunoliposome: immune cell-mediated anti-tumor activities. Cancer Lett 2005; 228(1–2): 181–186.
- Mitri Z, Constantine T, O’Regan R: The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract 2012; 2012: 743193.
- Slamon DJ, Godolphin W, Jones LA, et al.: Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244(4905): 707–712.
- Zhang JG, Kruse CA, Driggers L, et al.: Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol 2008; 88(1): 65–76.
- Casucci M, Hawkins RE, Dotti G, Bondanza A: Overcoming the toxicity hurdles of genetically targeted T cells. Cancer Immunol Immunother 2015; 64(1): 123–130.
- Morgan RA, Yang JC, Kitano M, et al.: Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4): 843–851.
- Slamon DJ, Leyland-Jones B, Shak S, et al.: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344(11): 783–792.
- Wang Y, Geldres C, Ferrone S, Dotti G: Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen receptor-based T-cell immunotherapy of solid tumors. Expert Opin Ther Targets 2015; 19(10): 1339–1350.
- Beard RE, Zheng Z, Lagisetty KH, et al.: Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer 2014; 2: 25.
- Pellegatta S, Savoldo B, Di IN, et al.: Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci Transl Med 2018; 10(430).
- Geldres C, Savoldo B, Hoyos V, et al.: T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res 2014; 20(4): 962–971.
- Abken H, Hombach A, Heuser C, Reinhold U.: A novel strategy in the elimination of disseminated melanoma cells: chimeric receptors endow T cells with tumor specificity. Recent Results Cancer Res 2001; 158: 249–264.
- Burns WR, Zhao Y, Frankel TL, et al.: A high molecular weight melanoma-associated antigen-specific chimeric antigen receptor redirects lymphocytes to target human melanomas. Cancer Res 2010; 70(8): 3027–3033.
- Losch FO, Muller R, Mutschler B, et al.: Activation of T cells via tumor antigen specific chimeric receptors: the role of the intracellular signaling domain. Int J Cancer 2003; 103(3): 399–407.
- Reinhold U, Liu L, Ludtke-Handjery HC, et al.: Specific lysis of melanoma cells by receptor grafted T cells is enhanced by anti-idiotypic monoclonal antibodies directed to the scFv domain of the receptor. J Invest Dermatol 1999; 112(5): 744–750.
- Schmidt P, Kopecky C, Hombach A, et al.: Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A 2011; 108(6): 2474-2479.
- Harrer D, Simon B, Fujii SI, et al.: RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer 2017; 17: 551.
- Bumol TF, Reisfeld RA.: Unique glycoprotein-proteoglycan complex defined by monoclonal antibody on human melanoma cells. Proc Natl Acad Sci USA 1982; 79(4): 1245–1249.
- Ilieva KM, Cheung A, Mele S, et al.: Chondroitin Sulfate Proteoglycan 4 and Its Potential As an Antibody Immunotherapy Target across Different Tumor Types. Front Immunol 2017; 8: 1911.
- Schiffer D, Mellai M, Boldorini R, et al.: The Significance of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Human Gliomas. Int J Mol Sci 2018; 19(9).
- Van Sinderen M, Cuman C, Winship A, et al.: The chrondroitin sulfate proteoglycan (CSPG4) regulates human trophoblast function. Placenta 2013; 34(10): 907–912.
- Fukushi J, Makagiansar IT, Stallcup WB: NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha3beta1 integrin. Mol Biol Cell 2004; 15(8): 3580–3590.
- Sakry D, Neitz A, Singh J, et al.: Oligodendrocyte precursor cells modulate the neuronal network by activity-dependent ectodomain cleavage of glial NG2. PLoS Biol 2014; 12(11): e1001993.
- Legg J, Jensen UB, Broad S, et al.: Role of melanoma chondroitin sulphate proteoglycan in patterning stem cells in human interfollicular epidermis. Development 2003; 130(24): 6049–6063.
- Ferrone S, Chen ZJ, Liu CC, et al.: Human high molecular weight-melanoma associated antigen mimicry by mouse anti-idiotypic monoclonal antibodies MK2-23. Experimental studies and clinical trials in patients with malignant melanoma. Pharmacol Ther 1993; 57(2-3): 259–290.
- Schlingemann RO, Rietveld FJ, de Waal RM, et al.: Expression of the high molecular weight melanoma-associated antigen by pericytes during angiogenesis in tumors and in healing wounds. Am J Pathol 1990; 136(6): 1393–1405.
- Midwood KS, Salter DM: Expression of NG2/human melanoma proteoglycan in human adult articular chondrocytes. Osteoarthritis Cartilage 1998; 6(5): 297–305.
- Tordsson JM, Ohlsson LG, Abrahmsen LB, et al.: Phage-selected primate antibodies fused to superantigens for immunotherapy of malignant melanoma. Cancer Immunol Immunother 2000; 48(12): 691–702.
- Petrini S, Tessa A, Carrozzo R, et al.: Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin-negative and Duchenne muscular dystrophies. Mol Cell Neurosci 2003; 23(2): 219–231.
- Campoli MR, Chang CC, Kageshita T, et al.: Human high molecular weight-melanoma-associated antigen (HMW-MAA): a melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit Rev Immunol 2004; 24(4): 267–296.
- Beard RE, Abate-Daga D, Rosati SF, et al.: Gene expression profiling using nanostring digital RNA counting to identify potential target antigens for melanoma immunotherapy. Clin Cancer Res 2013; 19(18): 4941–4950.
- Erfurt C, Sun Z, Haendle I, et al.: Tumor-reactive CD4+ T cell responses to the melanoma-associated chondroitin sulphate proteoglycan in melanoma patients and healthy individuals in the absence of autoimmunity. J Immunol 2007; 178(12): 7703–7709.
- Natali PG, Giacomini P, Russo C, et al.: Antigenic profile of human melanoma cells. Analysis with monoclonal antibodies to histocompatibility antigens and to melanoma-associated antigens. J Cutan Pathol 1983; 10(4): 225–237.
- Berd D, Herlyn M, Koprowski H, Mastrangelo MJ: Flow cytometric determination of the frequency and heterogeneity of expression of human melanoma-associated antigens. Cancer Res 1989; 49(23): 6840–6844.
- Morgan AC Jr., Galloway DR, Reisfeld RA: Production and characterization of monoclonal antibody to a melanoma specific glycoprotein. Hybridoma 1981; 1(1): 27–36.
- Morgan AC Jr., Woodhouse C, Bartholemew R, Schroff R: Human melanoma-associated antigens: analysis of antigenic heterogeneity by molecular, serologic and flow-cytometric approaches. Mol Immunol 1986; 23(2): 193–200.
- Li Y, Madigan MC, Lai K, et al.: Human uveal melanoma expresses NG2 immunoreactivity. Br J Ophthalmol 2003; 87(5): 629–632.
- Li Y, Wang J, Rizvi SM, Jager MJ, et al.: In vitro targeting of NG2 antigen by 213Bi-9.2.27 alpha-immunoconjugate induces cytotoxicity in human uveal melanoma cells. Invest Ophthalmol Vis Sci 2005; 46(12): 4365–4371.
- Chekenya M, Rooprai HK, Davies D, et al.: The NG2 chondroitin sulfate proteoglycan: role in malignant progression of human brain tumours. Int J Dev Neurosci 1999; 17(5–6): 421–435.
- Godal A, Bruland O, Haug E, et al.: Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br J Cancer 1986; 53(6): 839–841.
- Shoshan Y, Nishiyama A, Chang A, et al.: Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci U S A 1999; 96(18): 10361–10366.
- Yadavilli S, Hwang EI, Packer RJ, Nazarian J: The Role of NG2 Proteoglycan in Glioma. Transl Oncol 2016; 9(1): 57–63.
- Behm FG, Smith FO, Raimondi SC, et al.: Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood 1996; 87(3): 1134–1139.
- Hilden JM, Smith FO, Frestedt JL, et al.: MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 1997; 89(10): 3801–3805.
- Schwartz S, Rieder H, Schlager B, et al.: Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia 2003; 17(8): 1589–1595.
- Smith FO, Rauch C, Williams DE, et al.: The human homologue of rat NG2, a chondroitin sulfate proteoglycan, is not expressed on the cell surface of normal hematopoietic cells but is expressed by acute myeloid leukemia blasts from poor-prognosis patients with abnormalities of chromosome band 11q23. Blood 1996; 87(3): 1123–1133.
- Wuchter C, Harbott J, Schoch C, et al.: Detection of acute leukemia cells with mixed lineage leukemia (MLL) gene rearrangements by flow cytometry using monoclonal antibody 7.1. Leukemia 2000; 14(7): 1232–1238.
- Wang X, Osada T, Wang Y, et al.: CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst 2010; 102(19): 1496–1512.
- Nicolosi PA, Dallatomasina A, Perris R: Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015; 5(5): 530–544.
- de Vries JE, Keizer GD, te Velde AA, et al.: Characterization of melanoma-associated surface antigens involved in the adhesion and motility of human melanoma cells. Int J Cancer 1986; 38(4): 465–473.
- Ozerdem U: Targeting pericytes diminishes neovascularization in orthotopic uveal melanoma in nerve/glial antigen 2 proteoglycan knockout mouse. Ophthalmic Res 2006; 38(5): 251–254.
- Ozerdem U: Targeting of pericytes diminishes neovascularization and lymphangiogenesis in prostate cancer. Prostate 2006; 66(3): 294–304.
- Ampofo E, Schmitt BM, Menger MD, Laschke MW: The regulatory mechanisms of NG2/CSPG4 expression. Cell Mol Biol Lett 2017; 22: 4.
- Stoiber S, Cadilha BL, Benmebarek MR, et al.: Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019; 8(5).
- Sievers NM, Dörrie J, Schaft N: CARs: Beyond T Cells and T Cell-Derived Signaling Domains. Int J Mol Sci 2020; 21(10).
- Harrer DC, Dörrie J, Schaft N: Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum Gene Ther 2018; 29(5): 547–558.
- Montagner IM, Penna A, Fracasso G, et al.: Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells 2020; 9(6).
- Van Cutsem E, Machiels J, Van den Eynde M, et al.: SO-009 – Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Annals of Oncology 2019; 30: iv124–iv125.
- Santoro SP, Kim S, Motz GT, et al.: T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res 2015; 3(1): 68–84.
- Bansal R, Reshef R: Revving the CAR – Combination strategies to enhance CAR T cell effectiveness. Blood Reviews 2020: 100695.
- Harrer DC, Dorrie J, Schaft N: CARs and Drugs: Pharmacological Ways of Boosting CAR-T-Cell Therapy. Int J Mol Sci 2023; 24(3).
DERMATOLOGIE PRAXIS 2024; 19(1): 14–24