Le terme de lymphomes indolents englobe un nombre croissant de lymphomes non hodgkiniens de faible malignité et de leucémies, principalement de la lignée des cellules B. Les lymphomes indolents sont des tumeurs qui se développent à partir de cellules souches. Ils sont différenciés et classés les uns des autres en fonction de leur constellation de marqueurs et de leur profil génétique. Le traitement dépend de l’ampleur ou de la dynamique de la maladie et des symptômes du patient. On assiste actuellement à un glissement du traitement des chimiothérapies classiques vers les inhibiteurs de signal et les immunothérapies. L’objectif principal du traitement est souvent de contrôler la maladie le plus longtemps possible tout en maintenant une tolérance acceptable au traitement.
Le terme de lymphome non hodgkinien indolent (LNHi) désigne un groupe biologiquement hétérogène de lymphomes matures et à petites cellules de la lignée des cellules B ayant tendance à avoir des manifestations leucémiques. Auparavant, le terme “indolent” décrivait un groupe de lymphomes “de faible malignité” à croissance lente, dont les sous-types étaient souvent difficiles à séparer les uns des autres et qui ne devaient pas toujours être différenciés en raison du manque de possibilités thérapeutiques.

Entre-temps, des données immunophénotypiques et moléculaires élargies permettent une meilleure subdivision en entités de lymphome, qui devrait être effectuée selon la classification actuelle des lymphomes de l’OMS en 2008 (tab. 1) [1]. Certains LNHI se transforment en lymphomes blastiques ou agressifs hautement malins au cours de la maladie, le lymphome folliculaire en tête, suivi de la leucémie lymphoïde chronique (LLC). En outre, il existe des entités telles que le lymphome à cellules du manteau (MCL), dont la majorité (environ 90% de tous les cas) présentent un comportement de croissance agressif et sont traitées par immunochimiothérapie intensive (éventuellement avec une thérapie à haute dose et un remplacement de cellules souches autologues). Mais il existe aussi un sous-groupe indolent, qui se manifeste généralement chez les patients âgés présentant une atteinte de la moelle osseuse et de la rate. Il ne faut pas oublier les LNH typiques qui peuvent survenir dans un contexte clinique particulier, comme le MALT associé à Helicobacter pylori ou le lymphome de la zone marginale associé au VHC.
Diagnostic et sous-types
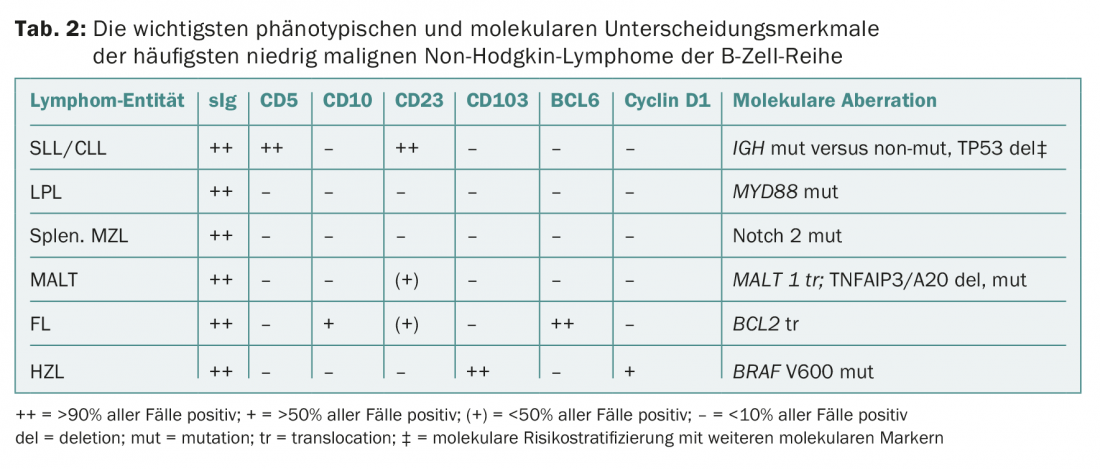
Le diagnostic est établi par une biopsie des ganglions lymphatiques et/ou de la moelle osseuse, un (immuno)phénotypage du sang et/ou de la moelle osseuse et des analyses génétiques et moléculaires (tableau 2). Ces tests sont standardisés et s’appliquent à toutes les pathologies du lymphome. Selon la classification de l’OMS, les entités suivantes font partie des iNHL :
- Leucémie lymphoïde chronique (LLC) ou leucémie à tricholeucocytes (LT) leur forme nodulaire (lymphome lymphocytaire à petites cellules, SLL)
- Lymphome lymphoplasmocytaire (LPL ou maladie de Waldenström)
- Lymphome de la zone marginale (LZM) (Fig. 1) avec ses trois sous-formes : nodale, extranodale (par ex. comme MALT) et splénique
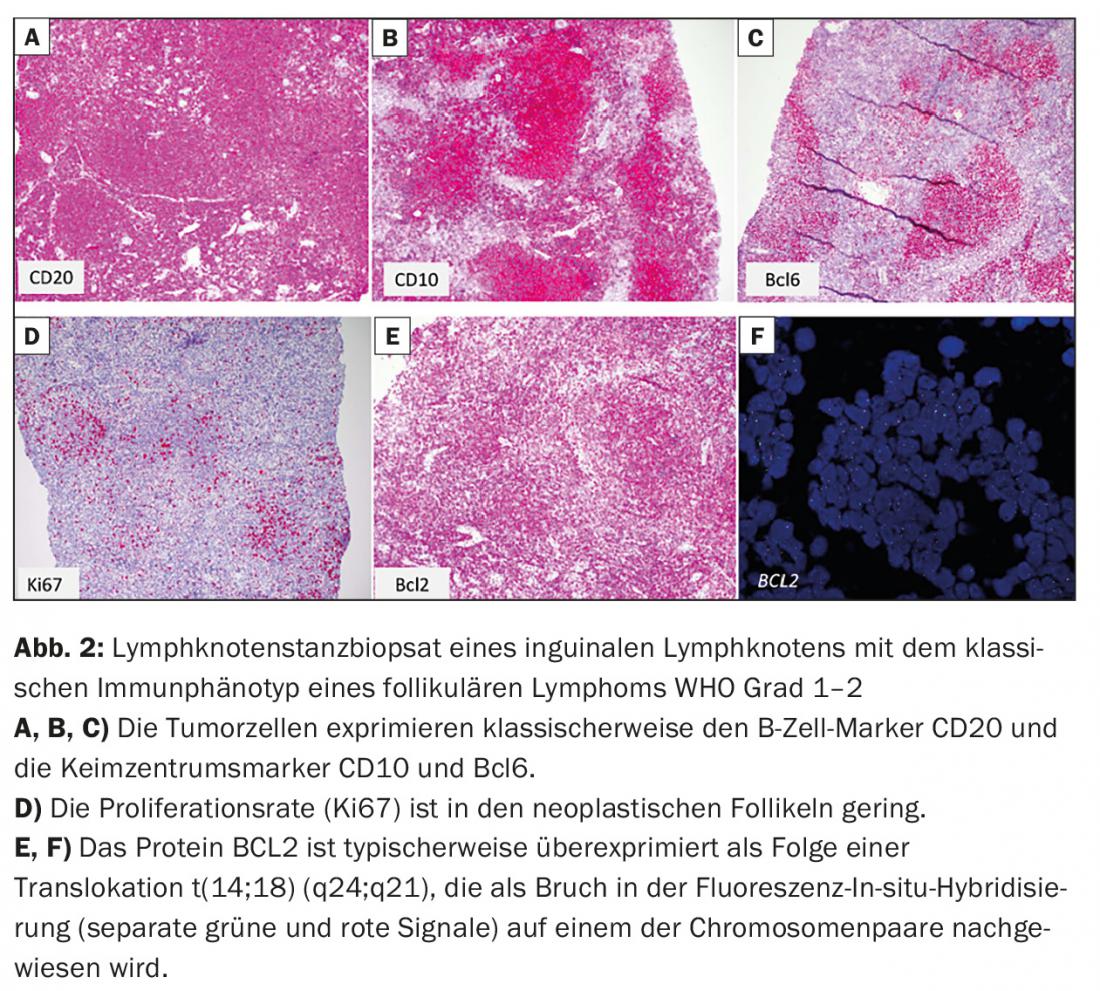
- Lymphome folliculaire (FL) (Fig. 2)
- La leucémie à tricholeucocytes (HZL).
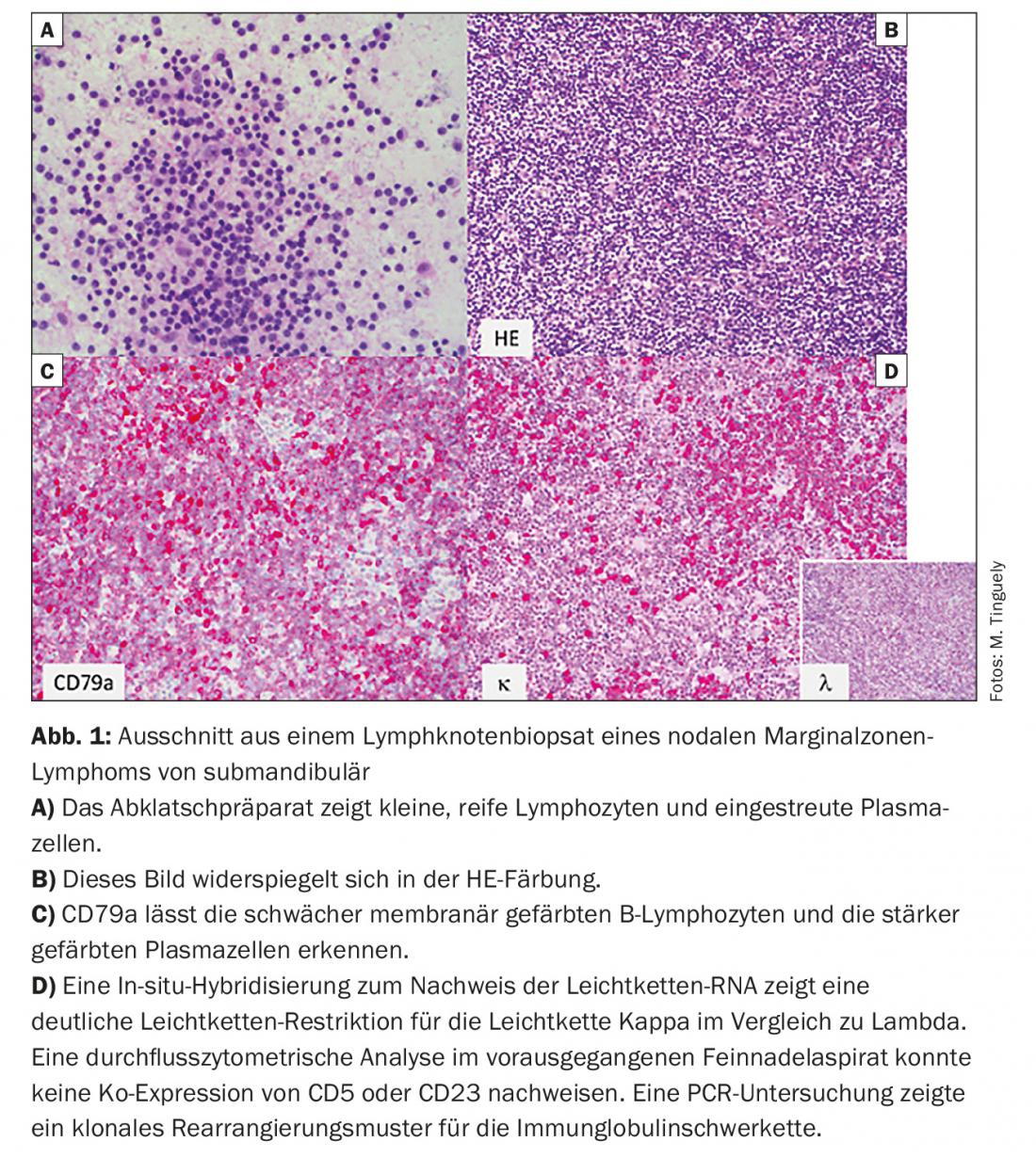
La LLC et la leucémie à tricholeucocytes ne sont pas considérées par tous les groupes de travail comme faisant partie du LNHI car, comme leur nom l’indique, elles se manifestent souvent en premier lieu par des modifications de la formule sanguine et nécessitent parfois (cela concerne en particulier la leucémie à tricholeucocytes) d’autres mesures thérapeutiques. Le lymphome à cellules du manteau (MCL) ne devrait plus être classé parmi les iNHL pour les raisons susmentionnées, bien qu’il existe un sous-groupe indolent chez les patients âgés.
Présentation clinique et principes thérapeutiques
Les lymphomes indolents sont généralement une maladie de l’âge avancé. La grande majorité des patients (66%) se trouvent déjà dans les stades avancés III et IV (classification des stades selon Ann Arbor ou plus récemment Lugano) au moment du diagnostic et ne sont donc plus accessibles à un traitement curatif [2]. L’établissement d’un diagnostic n’implique pas toujours la nécessité de mettre en place immédiatement un traitement. Même si les recommandations varient quelque peu selon les sous-types, il est toujours vrai que seules les maladies symptomatiques doivent être traitées. La définition d’une maladie symptomatique est restée largement inchangée au cours des 30 dernières années et inclut les symptômes locaux dus à la croissance du lymphome, à l’altération de la fonction normale des organes (par exemple, l’anémie symptomatique), les symptômes B, la maladie extranodale symptomatique, d’autres cytopénies ou une vitesse de croissance rapide d’une manifestation lymphatique. Ces critères sont très élastiques et offrent au patient et au médecin une grande marge de décision. C’est pourquoi des efforts sont actuellement déployés pour établir une meilleure classification pronostique et donc une aide à la décision au moyen de nouveaux marqueurs (moléculaires). L’indice pronostique de la LLC (CLL-IPI), récemment présenté, en est un exemple : il donne une recommandation quant au moment de commencer le traitement et au type de traitement [3].
La radiothérapie dans les stades précoces : une option curative ?
Au cours des dernières décennies, la radiothérapie a été définie comme une option de traitement curatif des premiers stades de la maladie du lymphome folliculaire. 15-25% des patients diagnostiqués avec un lymphome folliculaire sont au stade I ou II et les directives nationales et internationales en vigueur recommandent toujours la radiothérapie pour ces patients [4].
La difficulté d’une recommandation positive pour la mise en œuvre de la radiothérapie dans la pratique clinique quotidienne actuelle repose sur l’incertitude de savoir si les données des anciennes études (grandes surfaces d’irradiation, doses totales élevées, applications non spécifiques aux ganglions lymphatiques ou au champ d’involution) peuvent être appliquées aux techniques d’irradiation actuelles. Il existe donc un grand dilemme : nous ne pouvons actuellement pas définir la valeur curative présumée de la radiothérapie dans les lymphomes indolents (principalement le lymphome folliculaire) au stade précoce.
Attendre après le diagnostic : est-ce encore valable ?
Certains auteurs estiment qu’actuellement, environ 50% des patients n’ont toujours pas besoin d’un traitement immédiat au moment du diagnostic. Le terme “watch & wait” (w&w) a été inventé il y a plus de 35 ans [5]. L’attente semblait justifiée lorsque la maladie progressait lentement et que les symptômes étaient faibles ou inexistants – et aussi parce qu’il n’existait pas de traitement efficace. Ainsi, à l’époque, chez les patients FL, le délai médian avant le premier traitement était de 31 à 36 mois. Des analyses d’observation longitudinales ont montré qu’environ 20% des patients FL n’avaient pas besoin de traitement avec un suivi médian de 17 ans. Comparé à une cohorte de patients traités par chimiothérapie au moment du diagnostic, aucune différence n’a été observée en termes de survie globale (SG) à 5 ans. L’OS moyen était de 11 ans et variait considérablement d’une histologie à l’autre.
La question de savoir si le w&w est approprié concerne toutes les LNI, mais elle est principalement analysée dans le cas du FL. Ainsi, dans une étude multicentrique, 379 patients ont été randomisés dans trois bras de traitement : observation seule (w&w), induction par rituximab seul (applications toutes les quatre semaines) ou induction par rituximab suivie d’un traitement d’entretien de deux ans (rituximab tous les deux mois) [6]. L’étude a montré un avantage significatif en termes de survie sans progression (PFS) pour les deux bras de rituximab par rapport à w&w. L’étude a également montré que le rituximab n’avait pas d’effets secondaires. La survie globale n’était toutefois pas différente, de sorte qu’il n’y a toujours pas de raison impérieuse d’utiliser l’immunothérapie à un stade précoce. Cela peut changer avec les nouveaux médicaments (immunothérapeutiques) et devrait donc toujours être remis en question.
Thérapies actuelles
Les possibilités de traitement du LNHi ont radicalement changé ces dernières années avec l’établissement de nouvelles thérapies. Ils sont principalement basés sur les anticorps et l’inhibition de la tyrosine kinase (Fig. 3). Jusqu’à présent, l’immunothérapie avec des anticorps spécifiques du CD20 en monothérapie ou en combinaison avec des agents chimiothérapeutiques classiques domine le traitement de première ligne. Il faut toujours partir du principe qu’une guérison n’est pas possible malgré les méthodes thérapeutiques modernes ; la seule exception est la transplantation allogénique de cellules souches.
Anticorps monoclonaux spécifiques du CD20
L’exemple classique d’un anticorps spécifique du CD20 est le rituximab, qui fait désormais partie intégrante du traitement du lymphome à cellules B et peut être utilisé dans différentes entités, soit en monothérapie (par exemple FL), soit en association avec une chimiothérapie (par exemple CLL). Les nouveaux anticorps anti-CD20 (par exemple l’obinutuzumab) ont une toxicité cellulaire directe (activité ADCC) parfois encore plus élevée et peuvent ainsi éliminer plus efficacement les cellules du lymphome [7]. Dans le traitement de la LLC, par exemple, cela se traduit par une augmentation du nombre de patients présentant une maladie résiduelle minimale (MRD) négative. Cela signifie que chez les patients traités par obinutuzumab + chimiothérapie, la maladie – par rapport au traitement classique par rituximab + chimiothérapie – est plus souvent indétectable (38 contre 3% dans le sang périphérique), malgré des méthodes de détection sensibles. On ne sait pas encore si l’augmentation du taux de négativité de la MRD se traduira par une survie prolongée.
Combinaison d’anticorps et de chimiothérapie
Les anticorps spécifiques CD20 sont généralement utilisés en combinaison avec le chlorambucil, la bendamustine ou le CHOP. Le chlorambucil est souvent utilisé en combinaison avec l’obinutuzumab dans le traitement des patients âgés atteints de LLC ou le rituximab dans le traitement du lymphome MALT [8]. En revanche, la bendamustine, associée au rituximab, est considérée comme le standard de première ligne dans le lymphome folliculaire (grades 1 et 2) [9] et dans la LLC (avec des réserves sur la fludarabine-endoxan). Le rituximab avec CHOP est utilisé un peu moins souvent qu’auparavant et est principalement utilisé dans les variantes de lymphomes blastoïdes ou de grade FL 3 . En général, le nombre de cycles de chimiothérapie est souvent limité à six (toutes les trois à quatre semaines) et l’immunothérapie est administrée pendant la même durée ou encore en monothérapie pendant deux ans, dans le cadre d’un traitement d’entretien.
Immunomodulateurs
Les immunomodulateurs (IMID) sont de petites molécules (“small molecules”) qui sont généralement prises par voie orale. Les IMID comme le lénalidomide ont été utilisés pour la première fois avec succès dans le myélome multiple et sont désormais testés dans le lymphome avec des taux de réponse encourageants. L’association de lénalidomide et de rituximab chez les patients atteints de FL de grade 3 et de maladie récurrente ou réfractaire a permis d’obtenir un taux de réponse global (ORR) allant jusqu’à 86%. Lorsque la combinaison est utilisée directement dans le traitement de première ligne, des taux d’ORR allant jusqu’à 98% peuvent être atteints avec des taux élevés de rémission complète (RC) (87% de RC et de RC non confirmées) et de négativité de la MRD. Récemment, l’autorisation de mise sur le marché a également été obtenue pour le traitement du lymphome à cellules du manteau (MCL). L’autorisation de mise sur le marché est basée sur des études portant sur la maladie MCL récurrente ou réfractaire sur la base d’un ROR de 42-53% [10,11].
Inhibition de la voie de signalisation du récepteur des cellules B (BCR)
Des taux de réponse encore plus élevés dans le traitement du MCL peuvent être obtenus avec des substances qui inhibent la voie de signalisation en aval du BCR, par exemple les inhibiteurs de la tyrosine kinase de Bruton (BTK) ou de la kinase PI3 (PI3K) [12]. Ces deux kinases sont souvent actives de manière constitutive dans les cellules de lymphome et favorisent la prolifération ou la survie cellulaire.
Inhibiteurs de BTK
L’ibrutinib est le premier inhibiteur de BTK autorisé qui se lie de manière irréversible et covalente à un résidu cystéine (Cys-481) de la tyrosine kinase, provoquant ainsi une inhibition puissante et durable de l’activité enzymatique. Dans la LLC, l’ibrutinib a montré une activité élevée. L’autorisation de mise sur le marché repose sur une étude comparative avec l’ofatumumab, un anticorps monoclonal spécifique du CD20 (étude RESONATE), menée chez 391 patients atteints de LLC/SLL prétraités [13]. Avec un suivi médian de 9,4 mois, l’ibrutinib (420 mg/d/po) a significativement amélioré la PFS et l’OS. Après 12 mois, l’OS était de 90% dans le groupe ibrutinib et de 81% dans le groupe ofatumumab. Le taux de réponse global était significativement plus élevé pour l’ibrutinib (42,6 vs. 4,1%, p <0,001). Le taux et la durée de la réponse étaient indépendants de la présence d’une del17p ou d’une résistance aux analogues des purines. Cela prouve la grande importance des inhibiteurs de BTK dans ce sous-groupe de patients atteints de LLC difficile à traiter. Les événements indésirables les plus fréquents ont été la diarrhée, la fatigue, la fièvre et les nausées.
La deuxième entité autorisée en Suisse concerne le traitement du MCL récidivant. L’autorisation de mise sur le marché a été accordée sur la base d’une étude multicentrique de phase II à un seul bras portant sur 111 patients atteints de MCL prétraités et recevant une dose de 560 mg d’ibrutinib une fois par jour. La publication complète fait état d’un ORR de 66% avec un taux de RC de 17% pour une durée médiane de réponse (DOR) de 17,5 mois [4]. Il est intéressant de noter que le taux de réponse a augmenté de manière continue au cours du traitement (ce que l’on appelle “incremental response under treatment”), de sorte que – contrairement aux chimiothérapies classiques – des rémissions tardives peuvent également survenir si le traitement est poursuivi.
Inhibiteurs de PI3K
La famille PI3K est composée d’un certain nombre de sérine/thréonine kinases qui régulent la croissance, la différenciation, le métabolisme, la survie et la prolifération dans différentes cellules. Par exemple, l’inhibition de l’unité p110δ entraîne une déplétion significative des cellules B et un blocage de la voie de signalisation en aval du BCR. C’est pourquoi la plupart des approches thérapeutiques dans le traitement du lymphome se focalisent sur un blocage direct de l’unité p110δ. Le prototype de cette classe de substances est l’idélalisib en tant qu’inhibiteur sélectif de p110δ.
L’idelalisib a d’abord été testé dans une étude randomisée de phase III chez 220 patients atteints de LLC en récidive en association avec le rituximab [15]. Dans le bras de contrôle, les patients ont reçu du rituximab plus un placebo. Avec un prétraitement médian de trois substances, le rituximab et un alkylant ou un analogue de nucléotide pur avaient été utilisés au préalable dans presque tous les cas. Près de 40% des patients atteints de LLC présentaient également l’altération génétique défavorable du gène p53.
En termes d’efficacité, on a observé un taux de réponse significativement plus élevé dans le bras rituximab + idelalisib (81 vs 13%), une prolongation significative de la SSP à 1 an (93 vs 46%, p<0,001) et une prolongation significative de l’OS à 1 an (92 vs 80%, p=0,02). La supériorité de l’association rituximab + idelalisib a été démontrée pour tous les sous-groupes.
Les effets secondaires significatifs dans la pratique clinique quotidienne et dépassant le cadre de l’application du rituximab sont la diarrhée précoce ou, dans certains cas, tardive. Mais si l’on compare son profil d’effets secondaires à celui d’autres substances susceptibles d’être utilisées dans cette situation, comme l’ofatumumab, l’alemtuzumab ou les cytostatiques traditionnels, l’idélalisib obtient certainement un résultat positif.
En monothérapie, l’idélalisib s’avère très efficace dans le traitement des LNHi en rechute. La molécule est approuvée pour le traitement des patients FL avec une maladie récurrente ayant reçu deux lignes de traitement précédentes [16]. Dans l’étude de phase II à un bras sous-jacente portant sur 125 patients atteints de LNHi résistants au rituximab et aux alkylants, l’idélalisib a été administré à raison de 150 mg deux fois par jour jusqu’à la progression de la maladie. Le délai médian de réponse était de 1,9 mois, la durée médiane de réponse de 12,5 mois et la PFS médiane de 11 mois. Les effets secondaires de grade 3 ou plus les plus fréquents étaient la neutropénie (27% des patients), l’augmentation des aminotransférases (13%), la diarrhée (13%) et la pneumonie (7%).
Inhibiteurs de BCL-2
La protéine anti-apoptotique BCL-2 est surexprimée dans les cellules de lymphome et contribue à la résistance à la chimiothérapie. Les inhibiteurs sélectifs de BCL-2 administrés par voie orale, comme le vénétoclax, stoppent la prolifération des cellules lymphomateuses et entraînent ainsi des rémissions tumorales dans des modèles précliniques. Dans une première étude clinique portant sur 106 patients atteints de MCL (n=28), de lymphome folliculaire (n=29), de lymphome diffus à grandes cellules B (n=41) et d’autres sous-types de LNH (n=8), le vénétoclax en monothérapie a montré un profil de sécurité acceptable à la dose maximale tolérée de 1200 mg/j [17]. L’ORR était de 44% pour tous les sous-types, de 78% pour le MCL et de 38% pour le FL. La PFS médiane était de 10 à 14 mois. Les effets indésirables liés au traitement les plus fréquents (EI ≥20%) ont été les nausées (48%), la diarrhée (44%), la fatigue (41%), la diminution de l’appétit (21%) et les vomissements (21%). La survenue d’un syndrome de lyse tumorale est importante, mais elle est survenue chez deux patients sans conséquences cliniques.
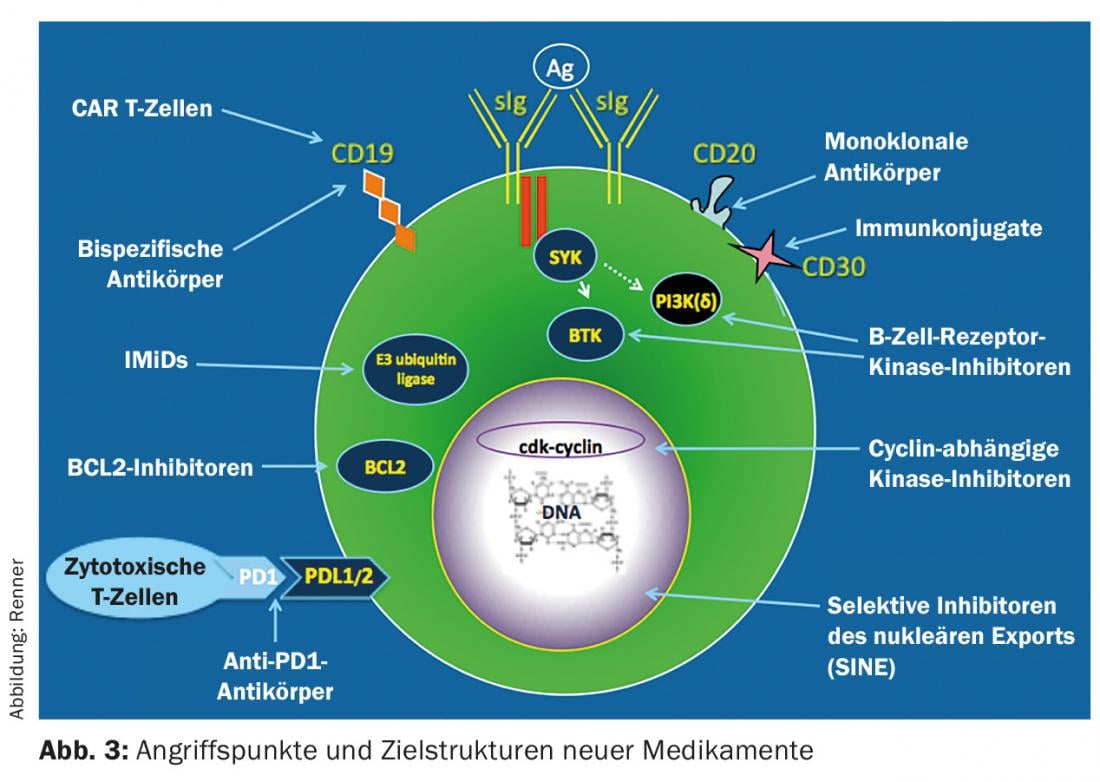
Futures thérapies
Les thérapies futures proviendront principalement de deux domaines (Fig. 3) : Les inhibiteurs de la voie de signalisation, qui ciblent des molécules de commutation importantes de la cellule lymphomateuse et bloquent leur fonction (par exemple, les inhibiteurs CDK 4/6), et les nouveaux médicaments d’immunothérapie. La découverte que le système immunitaire contribue au contrôle des tumeurs révolutionne actuellement les options de traitement hémato-oncologique. Les inhibiteurs de blocage des points de contrôle, les anticorps bispécifiques et les cellules T reprogrammées (CAR T cells) sont en cours de développement clinique, avec des résultats parfois impressionnants. Peut-être parviendrons-nous un jour à stimuler le système immunitaire de telle sorte qu’il sera possible de contrôler la tumeur à long terme, voire de la guérir.
Littérature :
- Swerdlow SH, et al. : IARC Press, Lyon, 2008.
- Brice P, et al : J Clin Oncol 1997 ; 15(3) : 1110-1117.
- Molica S, et al. : Abstract 498, présenté à la 57e réunion annuelle de l’Amercan Society of Hematology (ASH), 2015.
- Hiddemann W, et al. : Internist (Berl) 2016 ; 57(3) : 222-229.
- Morrison VA, Peterson BA : Leuk Lymphoma 1993 ; 10 Sup : 29-33.
- Adreshna KM, et al : Lancet Oncol 2014 ; 15(4) : 424-435.
- Goede V, et al. : NEJM 2014 ; 370(12) : 1101-1110.
- Zucca E, et al : J Clin Oncol 2013 ; 31(5) : 565-572.
- Rummel MJ, et al : Lancet 2013 ; 381(9873) : 1203-1210.
- Habermann TM, et al : Br J Haematol 2009 ; 145(3) : 344-349.
- Witzig TE, et al : Ann Oncol 2011 ; 22(7) : 1622-1627.
- Mato A, et al : Am J Hematol 2015 ; 90(7) : 657-664.
- Byrd JC, et al. : NEJM 2014 ; 371(3) : 213-223.
- Wang ML, et al. : NEJM 2013 ; 369(6) : 507-516.
- Furman RR, et al : NEJM 2014 ; 370(11) : 997-1007.
- Gopal AK, et al. : NEJM 2014 ; 370(11) : 1008-1018.
- Roberts AW, et al. : NEJM 2016 ; 374(4) : 311-322.
InFo ONKOLOGIE & HÄMATOLOGIE 2016 ; 4(2) : 11-15