
La sarcoïdose est une maladie granulomateuse multisystémique dont l’étiologie n’est pas encore claire. Le diagnostic de la sarcoïdose repose sur la présentation clinique typique et la mise en évidence histologique de granulomes non nécrosants à cellules épithélioïdes, après exclusion des autres diagnostics différentiels. Étant donné que plus de 90% des patients atteints de sarcoïdose ont des poumons ou des vaisseaux sanguins endommagés, il n’est pas nécessaire de les traiter. les ganglions lymphatiques intrathoraciques sont impliqués, le diagnostic est souvent établi à partir de biopsies obtenues par bronchoscopie. En cas d’indication, le traitement est effectué à l’aide d’immunosuppresseurs ou d’antiviraux. les immunomodulateurs.
La sarcoïdose est une maladie granulomateuse multisystémique dont l’étiologie n’est pas encore claire [1]. Elle se caractérise par des granulomes non nécrosants à cellules épithélioïdes dans différents organes et présente donc une clinique variable en fonction de l’organe atteint. Il n’existe pas de test de diagnostic spécifique, de sorte que le diagnostic repose sur des résultats cliniques typiques, la détection des granulomes typiques et l’exclusion des diagnostics différentiels.
La prévalence de la sarcoïdose est estimée à 1-40/100 000 habitants dans le monde, bien que les différentes définitions de cas, la présentation clinique variable et, en fin de compte, le diagnostic parfois difficile à poser rendent difficile un chiffrage précis [2,3]. L’étude ACCESS a confirmé que les Afro-Américains étaient plus souvent et plus gravement atteints [5].
En Suisse, une prévalence à vie de 121/100 000 a été constatée pour le diagnostic de sarcoïdose (130/100 000 hommes, 112/100 000 femmes). Une prévalence de 44/100 000 a été calculée pour la sarcoïdose active et de 16/100 000 pour la sarcoïdose nécessitant une hospitalisation. L’incidence annuelle moyenne en Suisse est estimée à 7/100 000 habitants [6].
La maladie peut survenir à tout âge, bien qu’elle soit plus fréquente chez les jeunes adultes (<40 ans) ; il existe un second pic de la maladie chez les femmes à l’âge de >50 ans [7]. En Suisse, l’âge moyen au moment du diagnostic est de 45 +/- 15 ans (41 +/- 14 ans pour les hommes, 48 +/- 15 ans pour les femmes) [6].
Cause et pathogenèse
Les causes de la sarcoïdose ne sont pas connues. Différentes hypothèses sont discutées dans la littérature. La maladie est probablement déclenchée par l’inhalation d’un agent/antigène (agent pathogène, aérosol) chez des patients génétiquement prédisposés, comme en témoigne l’observation d’une incidence accrue de la sarcoïdose au cours de la première année suivant l’attaque terroriste du World Trade Center à New York 9/11 chez les pompiers exposés à la poussière [8]. En Suisse, une prévalence accrue a été observée dans les régions où se trouvent des industries de transformation des métaux et dans les régions agricoles (transformation des pommes de terre et des céréales et exploitation des prairies) [6]. En raison de la caractéristique d’une maladie granulomateuse, l’exposition à des mycobactéries ou à d’autres agents pathogènes a également été évoquée, mais jamais prouvée.
L’observation d’une prévalence familiale et ethnique suggère qu’il existe un contexte génétique [9]. Différents allèles – principalement du complexe majeur d’histocompatibilité (CMH) et du gène ACE – ont été étudiés et des génotypes plus fréquemment associés à la sarcoïdose ont été identifiés [10]. Certains génotypes, quant à eux, ont été associés, entre autres, au syndrome de Löfgren et à une évolution bénigne [11].
Des processus immunologiques sont impliqués dans la pathogenèse de la sarcoïdose, mais ils ne sont pas encore compris en détail. La réponse immunitaire est supposée être déclenchée par la présentation d’un antigène non défini par les macrophages aux lymphocytes T CD4+ via les récepteurs CMH de classe II. Les chimiokines et cytokines (dont le TNF-α, l’IL-2) libérées à cette occasion entraînent la formation des granulomes typiques, composés de cellules épithélioïdes, de cellules géantes multinucléées et de lymphocytes T CD4+ [12–14]. On ne sait pas quels sont les mécanismes qui font que la maladie peut, d’une part, s’auto-limiter et, d’autre part, évoluer de manière chronique et progressive.
Diagnostic
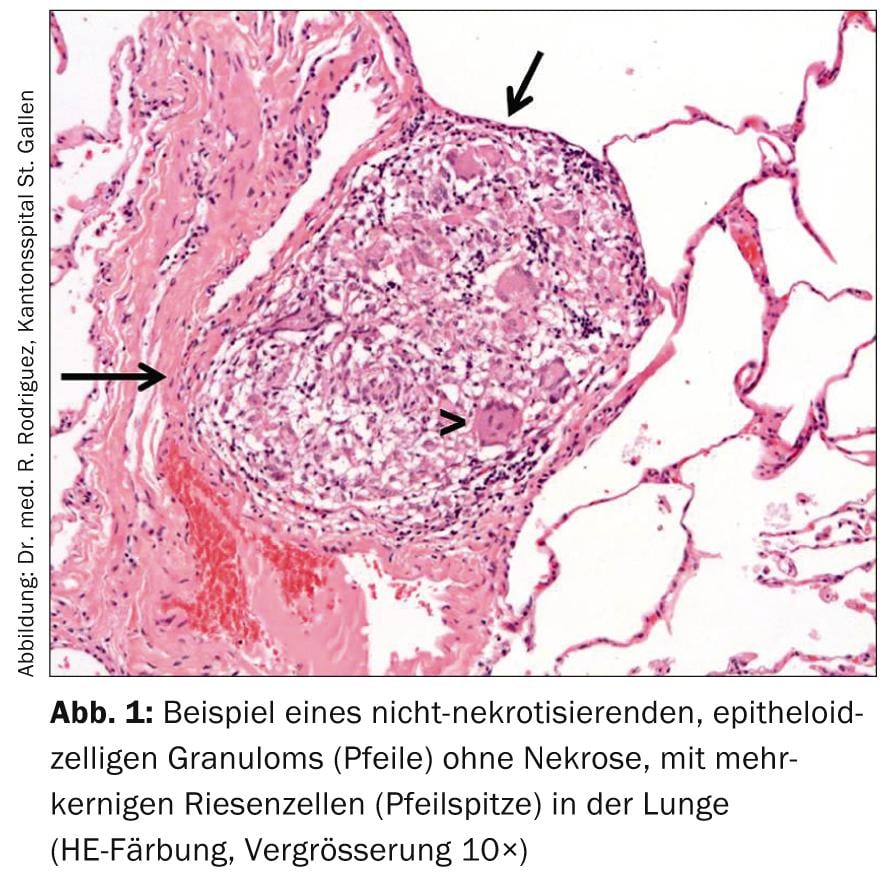
Le diagnostic de sarcoïdose est posé devant un tableau clinique typique, la mise en évidence histopathologique de granulomes non nécrosants à cellules épithélioïdes (Fig. 1) et après exclusion des diagnostics différentiels.
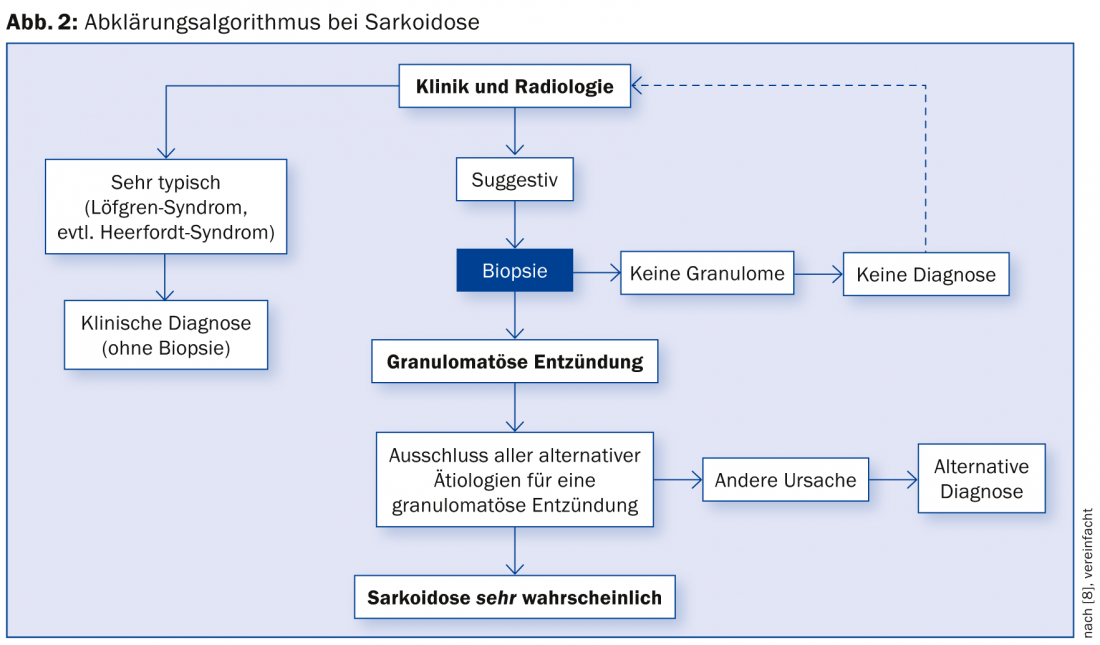
Un algorithme de diagnostic possible est résumé dans la figure 2 [15].
Symptômes typiques
Les granulomes typiques peuvent se développer dans n’importe quel organe. Les ganglions lymphatiques hilaires et médiastinaux, le tissu pulmonaire, la peau et les stations ganglionnaires extrathoraciques sont fréquemment touchés. Une implication ophtalmologique, neurologique ou cardiaque est moins souvent décrite (tab. 1) ; lorsque ces organes sont touchés, l’atteinte clinique, la morbidité et la mortalité sont considérables [16]. Il n’existe pas un seul symptôme typique de la sarcoïdose, car pratiquement tous les organes peuvent être touchés.

Souvent, les patients sont asymptomatiques, il n’est donc pas rare que la sarcoïdose soit une découverte fortuite lors d’une lymphadénopathie bihilique réalisée lors d’une radiographie conventionnelle [17].
Comme plus de 90% des patients présentent une manifestation pulmonaire, des symptômes tels qu’une toux non productive, une dyspnée et des douleurs thoraciques sont fréquents. Des symptômes généraux non spécifiques tels que fièvre, perte de poids, fatigue chronique, malaise, faiblesse musculaire et intolérance à l’effort sont également souvent décrits.
Syndrome de Löfgren et syndrome de Heerfordt
L’association d’une lymphadénopathie bihile, de fièvre, d’un érythème noueux et d’arthralgies/arthralgies est appelée syndrome de Löfgren. Cette clinique est caractéristique, ce qui permet de renoncer à la détection histologique des granulomes dans cette situation. Le syndrome de Löfgren a un bon pronostic. L’association rare mais spécifique d’une parotidite avec une uvéite et une parésie faciale périphérique (“Bells’ palsy”) avec des granulomes sarcoïdes histologiquement prouvés est connue sous le nom de syndrome de Heerfordt.
Manifestation intrathoracique
Les manifestations intrathoraciques vont d’une simple lymphadénopathie bihilique à une destruction sévère du parenchyme pulmonaire et à l’apparition d’une hypertension pulmonaire (même en l’absence de modifications pulmonaires). La gravité pulmonaire est divisée en cinq stades radiologiques selon Siltzbach et Scadding sur la base de la radiographie conventionnelle (tableau 2 et fig. 3) [18,19].

La classification par stade est importante, car elle permet de déduire des informations pronostiques. Deux tiers des patients présentent une rémission spontanée dans les 2 à 5 ans. Cela est particulièrement vrai pour les stades radiologiques I et II au moment du diagnostic.
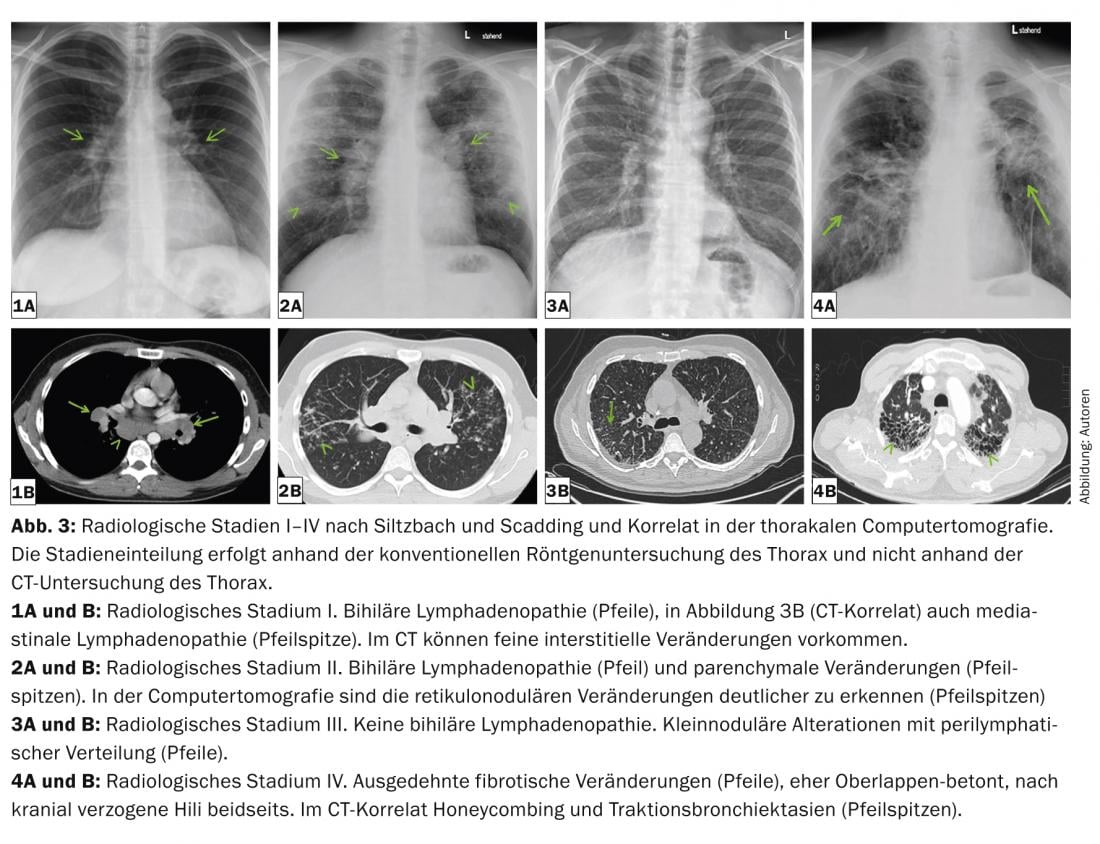
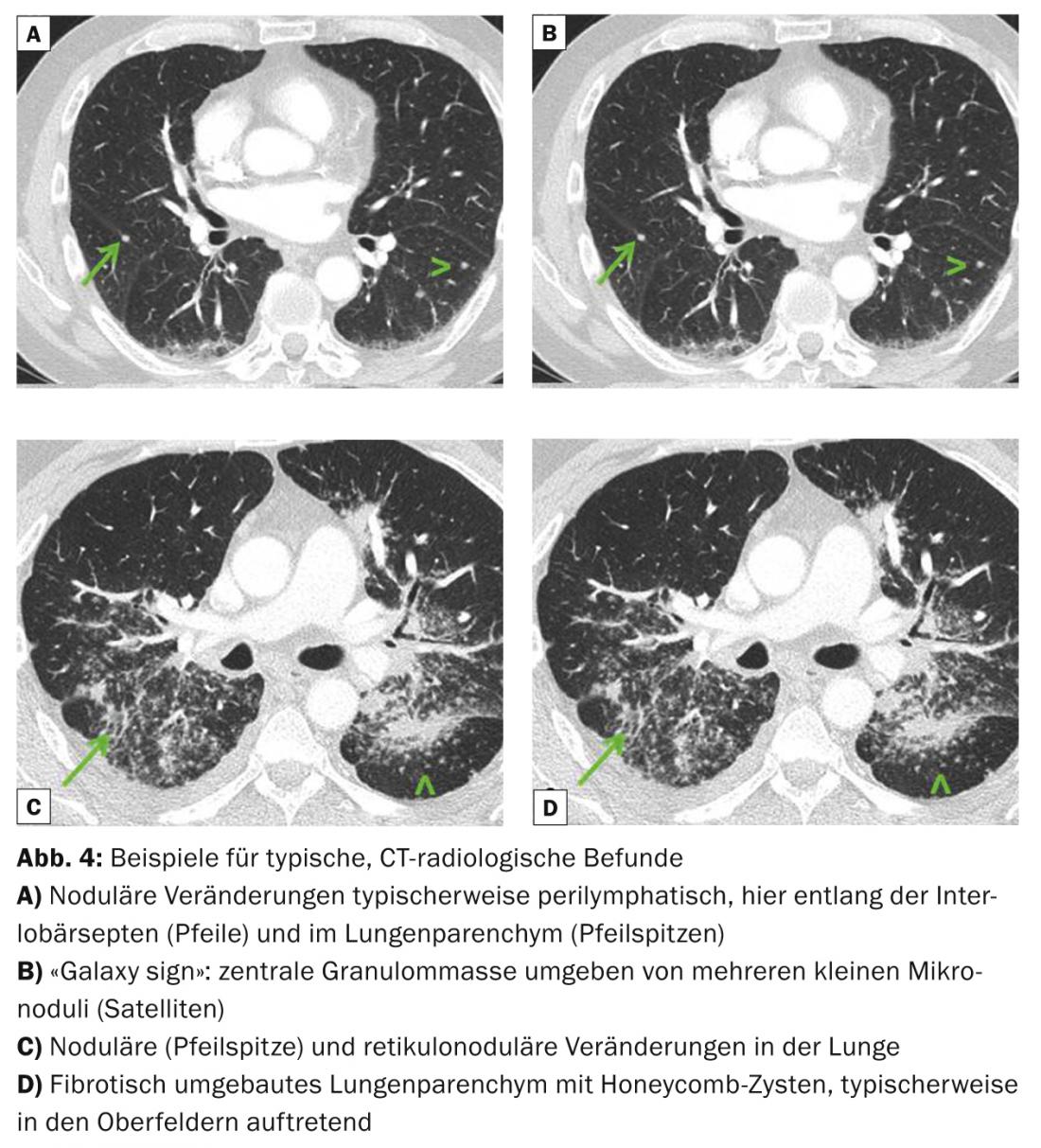
Les images radiologiques pulmonaires typiques sont une lymphadénopathie médiastinale bihilaire, mais aussi symétrique, des micronodules (diamètre <3 mm) avec une distribution typique (champs supérieur et moyen, le long des faisceaux bronchovasculaires, sous-pleuraux et le long des septa interlobulaires), des macronodules bilatéraux qui peuvent rarement fondre (reflétant une nécrose ischémique du noyau du granulome), des opacités en verre dépoli, une augmentation du dessin réticulaire, une fibrose avec honeycombing ainsi que des bronchectasies par traction avec accentuation des champs supérieurs. (Fig. 3 et 4). La tomodensitométrie thoracique (“High resolution computed tomography”, HRCT) est plus sensible pour l’évaluation de modifications discrètes du parenchyme pulmonaire.
Manifestations neurologiques, cardiaques ou ophtalmologiques
En cas de sarcoïdose cardiaque, il existe souvent des troubles de la conduction dans le sens d’un bloc AV ou de troubles de la conduction intraventriculaire sur l’ECG. Les patients peuvent être asymptomatiques, ressentir des palpitations ou des syncopes ou des crises d’épilepsie. subir des troubles du rythme pouvant aller jusqu’à la mort subite d’origine cardiaque. En cas d’implication grave du muscle cardiaque, il peut exister une cardiomyopathie avec un tableau clinique d’insuffisance cardiaque. En outre, une péricardite peut survenir.
La participation ophtalmologique se traduit souvent par une uvéite, une névrite du nerf optique et un gonflement des glandes lacrymales.
En cas d’implication neurologique, on observe le plus souvent une neuropathie crânienne (parésie faciale) ou périphérique, une irritation méningée avec détection de lymphocytes dans le LCR ou une infiltration de l’hypophyse ou de la glande pituitaire. de la région de l’hypothalamus, avec les complications endocrinologiques correspondantes.
L’apparition d’une hypercalcémie, conséquence d’une hydroxylation accrue de la vitamine D3 dans les cellules épithélioïdes des granulomes, peut s’avérer menaçante.
Manifestation cutanée
Différentes manifestations cutanées sont souvent décrites. Les symptômes typiques sont le lupus pernio et le lupus de l’intestin grêle. érythème noueux, mais aussi des lésions maculo-papuleuses et nodulaires (fig. 5) – souvent aussi dans la zone des tatouages et des cicatrices. Selon leur localisation (visage), les lésions cutanées peuvent entraîner une gêne subjective importante par défiguration.
Détection histologique des granulomes sarcoïdes
Sauf dans le cas du syndrome de Löfgren, il est recommandé d’établir le diagnostic par la mise en évidence histologique des granulomes typiques ; ceux-ci sont non nécrosants, petits, bien délimités avec une certaine accentuation périvasculaire et une tendance à l’agrégation, et éventuellement une fibrose centripète finement lamellaire. (Fig. 2). Le choix du site de biopsie doit permettre une approche aussi peu invasive que possible (par exemple, biopsie cutanée, biopsie de la glande lacrymale, excision de ganglions lymphatiques périphériques).
Seuls 2% environ des patients atteints de sarcoïdose extrapulmonaire n’ont pas d’atteinte pulmonaire [16]. C’est pourquoi les techniques de bronchoscopie sont utilisées pour tenter d’obtenir des résultats histologiques ou des données sur la maladie. de prélever des échantillons cytologiques. En cas de lésions endobronchiques (muqueuse modifiée en forme de pavé chez jusqu’à 50% des patients [20]), une biopsie de la muqueuse peut être obtenue à l’aide d’une pince ; un résultat positif peut être attendu chez plus de 60% des patients [21]. Les biopsies pulmonaires transbronchiques permettent d’obtenir des échantillons utilisables pour le diagnostic chez 60 à 97% des patients (en fonction du nombre de biopsies prélevées et de l’étendue des modifications radiologiques du parenchyme pulmonaire) [22–24], et les aspirations transbronchiques à l’aiguille (TBNA) guidées par échographie des ganglions lymphatiques médiastinaux et/ou hilaires chez environ 80% des patients [25]. La combinaison de ces techniques améliore le rendement diagnostique, de sorte que la médiastinoscopie, autrefois souvent nécessaire, n’est plus que très rarement utilisée.
Lors d’une bronchoscopie, un lavage broncho-alvéolaire est généralement effectué. Une sarcoïdose est probable – mais non prouvée – en cas de mise en évidence d’une alvéolite lymphocytaire (>15% de lymphocytes), d’un quotient CD4/CD8 élevé >3,5 et de l’exclusion d’une infection (entre autres mycobactéries). La sensibilité du quotient CD4/CD8 est de 42-59% et la spécificité de 76-96% pour l’augmentation du quotient >3,5 [26,27].
La technique PET/CT permet, dans des cas sélectionnés, de visualiser les foyers inflammatoires comme des régions où l’uptake du FDG est élevé. Si le site de la biopsie ne peut pas être clairement défini en raison des résultats cliniques, il est possible d’essayer d’identifier un foyer inflammatoire à l’aide de la technique PET/CT.
Diagnostics différentiels à exclure
Le diagnostic de sarcoïdose avec un tableau clinique typique et la mise en évidence d’un résultat histopathologique approprié peut être posé si d’autres maladies granulomateuses ont été exclues. Les diagnostics différentiels incluent les maladies infectieuses comme la tuberculose, les infections fongiques (histoplasmose, coccidioidomycose), la brucellose ou la tularémie. Ces maladies peuvent être exclues par le traitement du lavage broncho-alvéolaire en cas de suspicion clinique. Les diagnostics différentiels incluent également les maladies malignes, en particulier les lymphomes et les carcinomes (à exclure par l’examen histopathologique). La bérylliose chronique peut provoquer un tableau très proche de la sarcoïdose, notamment au niveau pulmonaire. Pour distinguer une bérylliose, il est important de recueillir une anamnèse professionnelle précise. Le diagnostic est établi, en cas de suspicion, par stimulation in vitro des cellules mononucléaires du sang ou du liquide du lavage broncho-alvéolaire. Il convient également d’exclure une alvéolite allergique exogène (antécédents, quotient CD4/CD8 abaissé), une pneumonie induite par des médicaments (antécédents) et des granulomes dans le cadre d’une réaction à un corps étranger (histologie). Comme les granulomes de la sarcoïdose peuvent également être périvasculaires, il peut être difficile de les distinguer d’une vascularite sur le matériel de biopsie.
De petits granulomes bien délimités dans les ganglions lymphatiques de drainage, dans le stroma environnant, mais aussi dans le foie ou la rate de patients atteints de maladies malignes sont appelés “sarcoid-like reaction” ; de tels granulomes apparaissent chez environ 4% des patients atteints de carcinomes (y compris le cancer du sein et les carcinomes rénaux et gastro-intestinaux) et un peu plus fréquemment chez les patients atteints de tumeurs malignes hodgkiniennes (14%) et non hodgkiniennes (7%). [28].
Un autre diagnostic différentiel important en cas de détection d’une maladie granulomateuse est le “Common variable immunodeficiency” (CVID). Elle se caractérise par de faibles taux d’immunoglobulines sériques, des infections bactériennes récurrentes et une diminution de la réponse en anticorps. Certains patients développent une inflammation granulomateuse avec des granulomes non nécrosants dans les poumons, la rate, le foie et les ganglions lymphatiques, entre autres [29].
La sarcoïdose est traitée, entre autres, par des inhibiteurs du TNF-α. Paradoxalement, des inflammations granulomateuses ont été décrites sous traitement par inhibiteurs du TNF-α, notamment sous étanercept dans la polyarthrite rhumatoïde, mais aussi sous infliximab [30]. Des granulomes non caséeux peuvent également apparaître dans le cadre d’un traitement à l’interféron γ (notamment en cas d’hépatite C chronique) [31].
Work-up et évaluation de l’évolution
Une fois le diagnostic de sarcoïdose posé, il est recommandé d’effectuer un bilan de l’étendue de la maladie. Il convient notamment de dépister les manifestations organiques susceptibles d’entraîner une morbidité importante (tableau 3).
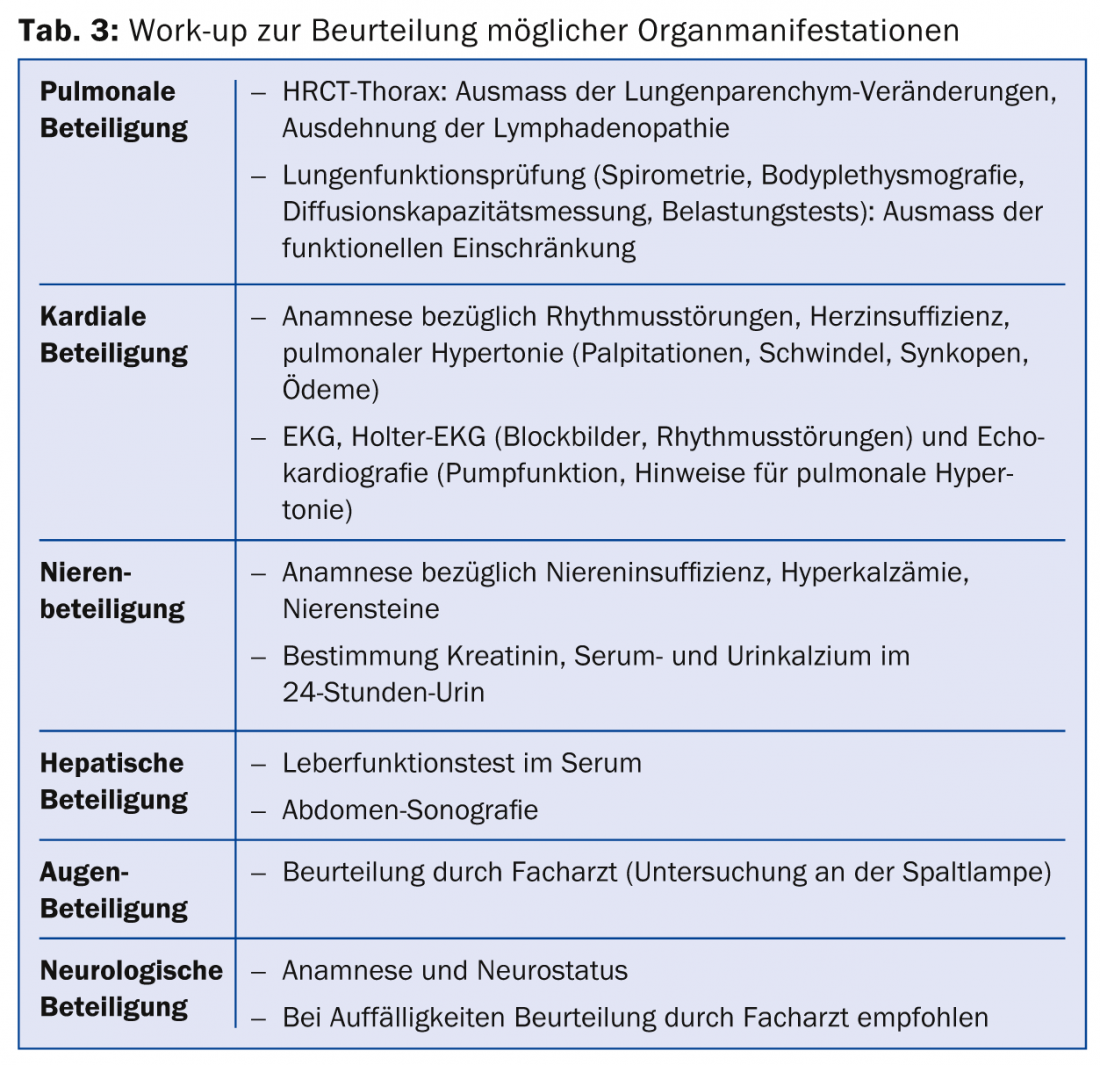
Il convient également de définir les paramètres qui peuvent être utilisés pour évaluer l’évolution (par exemple, même sous traitement). L’enzyme de conversion de l’angiotensine (ECA) est dosée pour évaluer une éventuelle participation médullaire. L’ECA est notamment produit par les cellules épithélioïdes des granulomes dans la sarcoïdose [32]. Cependant, en raison de sa faible sensibilité, l’ECA ne peut pas être utilisé comme test de diagnostic. Si elle est élevée au moment du diagnostic, l’ECA est un paramètre d’évolution possible. Le récepteur soluble de l’interleukine 2 est un autre paramètre évolutif possible : les cellules présentatrices d’antigènes produisent de l’interleukine 2 (IL-2) en relation avec la formation du granulome. Cela entraîne l’activation des lymphocytes T. Une forme soluble du récepteur de l’interleukine 2 (récepteur sIL-2) est alors libérée dans la circulation sanguine. Comme l’augmentation du récepteur sIL-2 reflète l’activation non spécifique des lymphocytes T, elle permet de déterminer l’activité de la maladie et donc d’évaluer son évolution.
Pronostic et indication de traitement
L’évolution de la maladie ne peut pas être prédite. Le pronostic est toutefois favorable chez environ deux tiers des patients : une rémission est observée dans la décennie, et chez environ 50% des patients dans les trois ans. Chez environ un tiers des patients, la maladie évolue de manière chronique et progressive, avec une limitation fonctionnelle importante des organes concernés. Moins de 5% des patients meurent de la sarcoïdose. La mortalité est principalement due à une fibrose pulmonaire avec insuffisance respiratoire, à une atteinte cardiaque ou neurologique ou à une hypertension pulmonaire [33].
L’indication de traitement doit être posée très soigneusement, en sachant que l’évolution de la maladie est généralement spontanément favorable, que les effets secondaires potentiels du traitement et que le taux de récidive est plus élevé après un traitement par immunosuppresseurs (l’antigène postulé persiste dans l’organe concerné, de sorte qu’une nouvelle formation de granulome peut survenir après l’arrêt du traitement). Il existe une indication thérapeutique absolue en cas de neurosarcoïdose, de sarcoïdose cardiaque ou oculaire avec atteinte du segment moyen ou postérieur de l’œil, ainsi qu’en cas d’atteinte fonctionnelle sévère des organes concernés, typiquement une insuffisance hépatique ou rénale, mais aussi une atteinte fonctionnelle pulmonaire sévère. En cas de limitation légère ou modérée, une surveillance clinique attentive de l’évolution est possible : en cas de symptômes existants et de progression de la limitation fonctionnelle, et donc d’indices d’activité de la maladie, un traitement est généralement mis en place.
Traitement par immunosuppresseurs/immunomodulateurs
S’il existe une indication de traitement (tab. 4), des immunosuppresseurs ou des médicaments à base de plantes sont administrés. immunomodulateurs sont utilisés (tableau 5) .
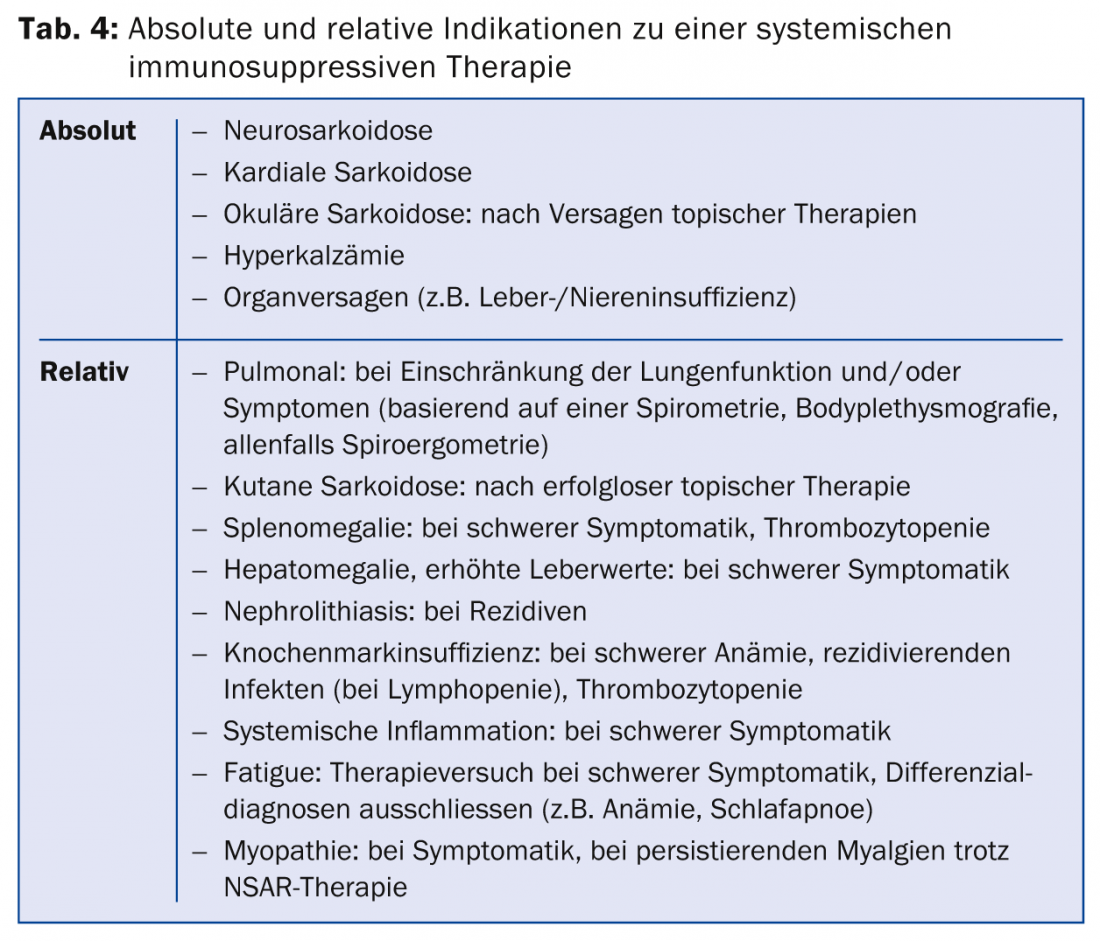
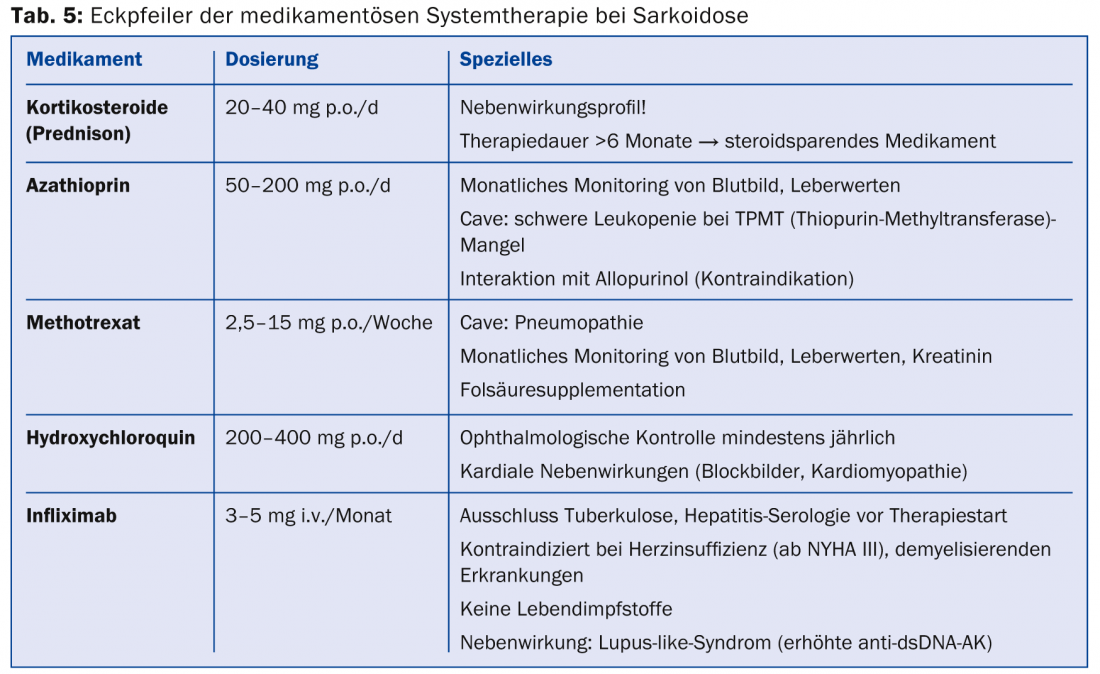
En raison de leur bonne et rapide efficacité, les corticostéroïdes sont des médicaments de premier choix. Des doses de 20-40 mg à doses progressives pendant environ six mois sont recommandées, mais il n’existe pas d’études randomisées contrôlées à ce sujet. L’utilisation de substances épargnant les stéroïdes (azathioprine, méthotrexate, léflunomide, mycophénolate) en cas d’effets secondaires associés aux stéroïdes doit être évaluée rapidement. Le méthotrexate doit cependant être prescrit avec réserve en cas de sarcoïdose pulmonaire en raison du risque de pneumonie au méthotrexate. Comme le TNF-α est impliqué dans la formation des granulomes, les antagonistes du TNF-α (surtout l’infliximab) sont une bonne alternative de traitement de seconde ligne [34,35]. Un bénéfice a été démontré en particulier pour le traitement de la sarcoïdose cutanée (lupus pernio) et des manifestations pulmonaires et neurologiques. L’hydroxychloroquine, un antipaludéen, donne de bons résultats dans la sarcoïdose cutanée et l’hypercalcémie. Dans le cas du syndrome de Löfgren, le traitement symptomatique par des anti-inflammatoires non stéroïdiens est bien établi.
Littérature :
- Hunninghake GW, et al. : Déclaration de l’ATS/ERS/WASOG sur la sarcoïdose. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis 1999 ; 16(2) : 149-173.
- Fernandez FE : [Epidemiology of sarcoidosis]. Arch Bronconeumol 2007 ; 43(2) : 92-100.
- Rybicki BA, Iannuzzi MC : Epidemiology of sarcoidosis : recent advances and future prospects. Semin Respir Crit Care Med 2007 ; 28(1) : 22-35.
- Pietinalho A, et al. : La fréquence de la sarcoïdose en Finlande et à Hokkaido, Japon. Une étude épidémiologique comparative. Sarcoidosis 1995 ; 12(1) : 61-67.
- Rossman MD, Kreider ME : Leçons tirées d’ACCESS (A Case Controlled Etiologic Study of Sarcoidosis). Proc Am Thorac Soc 2007 ; 4(5) : 453-456.
- Deubelbeiss U, et al : Prevalence of sarcoidosis in Switzerland is associated with environmental factors. Eur Respir J 2010 ; 35(5) : 1088-1097.
- Hosoda Y, et al. : Épidémiologie mondiale de la sarcoïdose. Quelle histoire nous racontent la prévalence et l’incidence ? Clin Chest Med 1997 ; 18(4) : 681-694.
- Izbicki G, et al : World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest 2007 ; 131(5) : 1414-1423.
- McGrath DS, et al : Epidémiologie de la sarcoïdose familiale au Royaume-Uni. Thorax 2000 ; 55(9) : 751-754.
- Rossman MD, et al : HLA-DRB1*1101 : a significant risk factor for sarcoidosis in blacks and whites. Am J Hum Genet 2003 ; 73(4) : 720-735.
- Berlin M, et al : HLA-DR predicts the prognosis in Scandinavian patients with pulmonary sarcoidosis. Am J Respir Crit Care Med 1997 ; 156(5) : 1601-1605.
- Baughman RP, et al. : Libération du facteur de nécrose tumorale par les macrophages alvéolaires de patients atteints de sarcoïdose. J Lab Clin Med 1990 ; 115(1) : 36-42.
- Iida K, et al. : Analyse des sous-ensembles de cellules T et des chimiokines bêta chez les patients atteints de sarcoïdose pulmonaire. Thorax 1997 ; 52(5) : 431-437.
- Pinkston P, Bitterman PB, Crystal RG : Libération spontanée d’interleukine-2 par les lymphocytes T pulmonaires dans la sarcoïdose pulmonaire active. N Engl J Med 1983 ; 308(14) : 793-800.
- Judson MA : Le diagnostic de la sarcoïdose. Clin Chest Med 2008 ; 29(3) : 415-427, viii.
- Baughman RP, et al : Caractéristiques cliniques des patients dans une étude de cas de sarcoïdose. Am J Respir Crit Care Med 2001 ; 164(10 Pt 1) : 1885-1889.
- Thomas KW, Hunninghake GW : Sarcoïdose. JAMA 2003 ; 289(24) : 3300-3303.
- Siltzbach LE : Sarcoïdose : caractéristiques cliniques et prise en charge. Med Clin North Am 1967 ; 51(2) : 483-502.
- Scadding JG : Pronostic de la sarcoïdose intrathoracique en Angleterre. Un examen de 136 cas après cinq ans d’observation. Br Med J 1961 ; 2(5261) : 1165-1172.
- Chapman JT, Mehta AC : Bronchoscopy in sarcoidosis : diagnostic and therapeutic interventions. Curr Opin Pulm Med 2003 ; 9(5) : 402-407.
- Shorr AF, Torrington KG, Hnatiuk OW. Biopsie endobronchique pour la sarcoïdose : une étude prospective. Chest 2001 ; 120(1) : 109-114.
- Koerner SK, et al : Biopsie pulmonaire transbronchique pour le diagnostic de la sarcoïdose. N Engl J Med 1975 ; 293(6) : 268-270.
- Gilman MJ, Wang KP : Biopsie pulmonaire transbronchique dans la sarcoïdose. Une approche pour déterminer le nombre optimal de biopsies. Am Rev Respir Dis 1980 ; 122(5) : 721-724.
- Mitchell DM, et al : Transbronchial lung biopsy through fibreoptic bronchoscope in diagnosis of sarcoidosis. Br Med J 1980 ; 280(6215) : 679-681.
- Agarwal R, et al : Efficacité et sécurité de la sonde convexe EBUS-TBNA dans la sarcoïdose : une revue systématique et une méta-analyse. Respir Med 2012 ; 106(6) : 883-892.
- Costabel U : CD4/CD8 ratios in bronchoalveolar lavage fluid : of value for diagnosing sarcoidosis ? Eur Respir J 1997 ; 10(12) : 2699-2700.
- Zaiss AW, et al : [T4/T8 ratio in bronchoalveolar lavage fluid : sensitivity and specificity for the diagnosis of sarcoidosis]. Prax Klin Pneumol 1988 ; 42 Suppl 1 : 233-234.
- Chowdhury FU, et al : Sarcoid-like reaction to malignancy on whole-body integrated (18)F-FDG PET/CT : prevalence and disease pattern. Clin Radiol 2009 ; 64(7) : 675-681.
- Ardeniz O, Cunningham-Rundles C : Maladie granulomateuse dans l’immunodéficience variable commune. Clin Immunol 2009 ; 133(2) : 198-207.
- Khasnis AA, Calabrese LH : Inhibiteurs du facteur de nécrose tumorale et maladie pulmonaire : un paradoxe d’efficacité et de risque. Semin Arthritis Rheum 2010 ; 40(2) : 147-163.
- Goldberg HJ, et al : Sarcoïdose après traitement par interféron-alpha : une série de cas et une revue de la littérature. Respir Med 2006 ; 100(11) : 2063-2068.
- Studdy PR, Bird R : L’enzyme de conversion de l’angiotensine sérique dans la sarcoïdose – sa valeur dans la pratique clinique actuelle. Ann Clin Biochem 1989 ; 26(Pt 1) : 13-18.
- Siltzbach LE, et al : Course and prognosis of sarcoidosis around the world. Am J Med 1974 ; 57(6) : 847-852.
- Judson MA, et al : Efficacité de l’infliximab dans la sarcoïdose extrapulmonaire : résultats d’un essai randomisé. Eur Respir J 2008 ; 31(6) : 1189-1196.
- Baughman RP, et al : Traitement par infliximab chez les patients atteints de sarcoïdose chronique et d’atteinte pulmonaire. Am J Respir Crit Care Med 2006 ; 174(7) : 795-802.
PRATIQUE DU MÉDECIN DE FAMILLE 2015 ; 10(3) : 10-18









