Les causes des plaies chroniques sont très variées et parfois rares. Un diagnostic correct est essentiel pour un traitement adéquat. Une biopsie est souvent utile lorsque les causes sont rares et peu claires. Le diagnostic et le traitement des plaies doivent être réalisés dans le cadre d’une prise en charge collaborative multimodale.
Joachim Dissemond, de l’hôpital universitaire d’Essen (Allemagne), dans le cadre de la réunion annuelle de l’Austrian Wound Association (AWA). “Le diagnostic et le traitement doivent absolument être interdisciplinaires et interprofessionnels”, appelle le conférencier, soulignant que la biopsie est très importante pour diagnostiquer les causes rares de plaies [1]. Afin d’identifier les facteurs déclencheurs ou une implication extracutanée, une anamnèse complète et des examens de laboratoire sélectionnés font également partie de la procédure d’investigation diagnostique. Les pathologies rares classées comme maladies orphelines sont décrites dans la base de données Orphanet [2].
Afin d’améliorer la qualité de vie des patients souffrant de plaies chroniques et de prévenir les complications liées aux plaies non cicatrisées, une prise en charge moderne des plaies et, le cas échéant, un traitement de la douleur sont indiqués comme mesures d’accompagnement.
Angiite leucocytoclasique cutanée – purpura palpable comme symptôme principal
L’angiite leucocytoclasique cutanée est la forme la plus courante de vascularite cutanée. La maladie peut survenir à tout âge et se manifeste typiquement par un purpura et/ou des pétéchies palpables sur les membres inférieurs avec une distribution unifocale ou multifocale. La vascularite est associée à une inflammation neutrophilique principalement limitée aux veinules post-capillaires cutanées superficielles et ne présente pas de vascularite ou de glomérulonéphrite systémique.
Des érythèmes livide apparaissent souvent, ce qui indique la présence d’une inflammation, explique le professeur Dissemond. Il en résulte des nécroses et, en règle générale, des ulcères multiples. La plupart du temps, les deux jambes sont touchées. “Une approche purement unilatérale serait extrêmement inhabituelle pour l’angiite leucocytoclasique cutanée”, a déclaré le conférencier [1].
Le diagnostic d’angiite leucocytoclasique cutanée est un diagnostic d’exclusion. En général, les échantillons de biopsie cutanée provenant de lésions datant de 24 à 48 heures doivent être examinés par microscopie optique et immunofluorescence directe. Les causes fréquentes de cette maladie rare sont les infections et/ou les médicaments, bien que jusqu’à 50% des cas soient idiopathiques. Le professeur Dissemond cite comme option de traitement classique les corticostéroïdes systémiques, par exemple la prednisolone 1 mg par kg/KG.
Pyoderma gangraenosum – Le score de Paracelse facilite le diagnostic
Le pyoderma gangraenosum se caractérise par des ulcères cutanés récurrents avec un exsudat muco-purulent ou hémorragique. Souvent, cela commence par une pustule stérile. Le pus est stérile parce qu’il s’agit de granulocytes neutrophiles, explique le professeur Dissemond [1]. L’incidence est la plus élevée dans le groupe d’âge des 20-50 ans, les femmes étant plus souvent touchées que les hommes. Les caractéristiques typiques de cette dermatose neutrophilique sont des bords sous-minés et un érythème violet sombre. Environ 70% des patients sont touchés sur la face d’extension des membres inférieurs (tibia), mais d’autres localisations sont possibles. Les comorbidités fréquentes sont les maladies inflammatoires chroniques de l’intestin (maladie de Crohn, colite ulcéreuse), mais les néoplasies ne sont pas rares chez ces patients. “C’est une maladie potentiellement paranéoplasique”, a déclaré le conférencier [1]. Par conséquent, les patients atteints de pyoderma gangraenosum, en particulier ceux dont l’évolution est réfractaire, doivent être examinés en particulier à la recherche de néoplasies hématologiques (par exemple, le syndrome myélodysplasique). Comment diagnostiquer un pyoderma gangraenosum ? La classification comme diagnostic d’exclusion n’est plus d’actualité, souligne le professeur Dissemond, qui ajoute : “Vous pouvez aujourd’hui poser le diagnostic avec relativement peu de mesures” [1]. L’utilisation du score de Paracelse, recommandé dans les directives allemandes, est utile à cet égard (tableau 1). Parmi les mesures thérapeutiques les plus importantes, le conférencier cite l’immunosuppression. Le traitement systémique par corticostéroïdes et ciclosporine A est le mieux documenté.

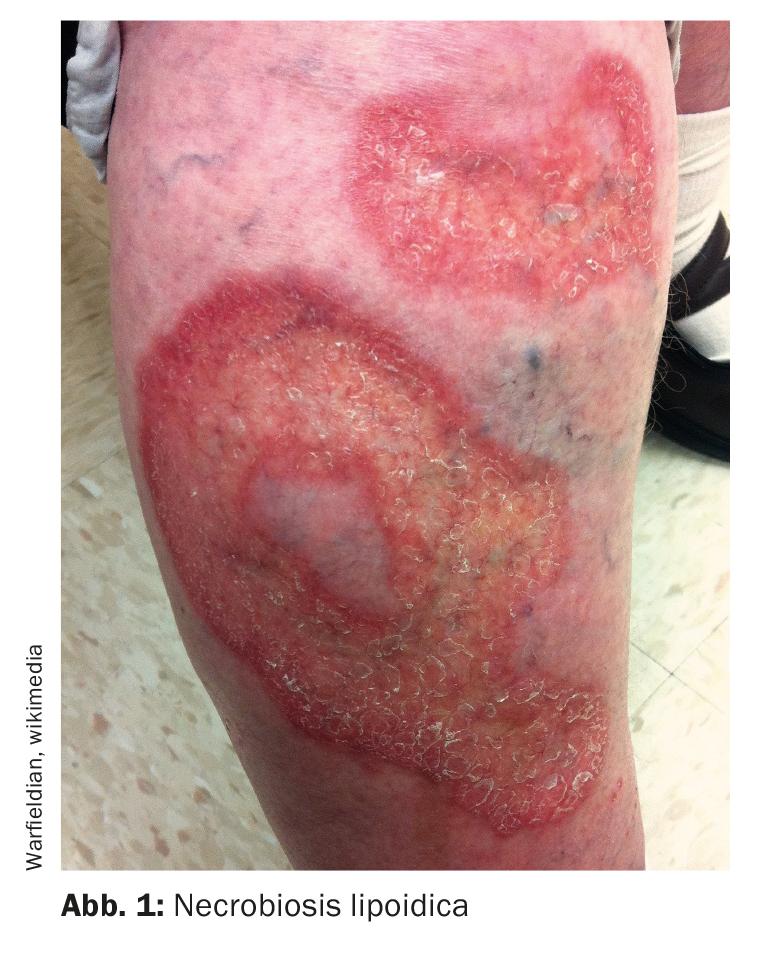
Necrobiosis lipoidica – les ulcérations sont extrêmement douloureuses
La nécrobiose lipoïdique (Fig. 1) est une maladie granulomateuse rare dont l’étiologie n’est pas encore bien comprise. Le site de prédilection de cette maladie orpheline est également le côté de l’extension des membres inférieurs – dans jusqu’à 90% des cas, les manifestations de la maladie sont localisées dans cette zone. Un signe clinique typique est la plaque jaune-orange avec atrophie et télangiectasies. “Ça ne s’ulcère pas forcément, mais quand ça s’ulcère, ça fait terriblement mal”, explique le professeur Dissemond [1]. En revanche, en l’absence d’ulcération, la nécrobiose lipoïdique est à peine perceptible par les patients. Cela commence généralement par une jambe, la deuxième jambe suivant après quelques mois ou années. Une biopsie est très importante pour le diagnostic. L’ancien nom était Necrobiosis lipoidica diabeticorum. Or, comme on le sait aujourd’hui, il n’y a association avec le diabète sucré que dans 50% des cas, explique le conférencier. Les personnes concernées ont toutefois un risque accru de développer un diabète.
Les possibilités de traitement se déroulent dans le domaine off-label. “Il n’existe pas de traitement approuvé”, a déclaré le conférencier [1]. Selon lui, le traitement par la cortisone appliquée par voie systémique est délicat chez les diabétiques. Le professeur Dissemond ajoute que l’on trouve dans la littérature des descriptions de l’utilisation de l’acide fumarique, et que certains produits biologiques (par exemple les inhibiteurs du TNF-alpha) sont également utilisés.
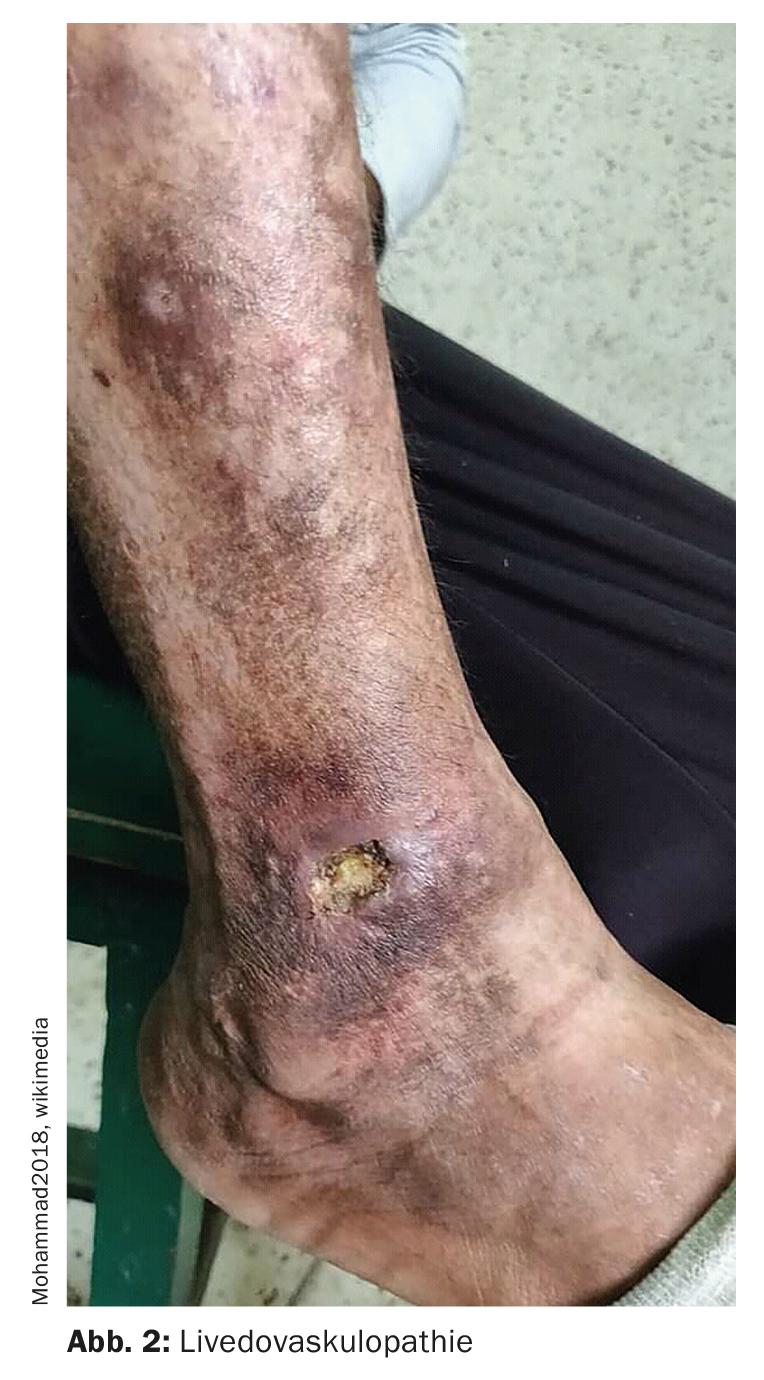
Livedovasculopathie – associée à des paramètres procoagulatoires
La livedovasculopathie (Fig. 2)est une maladie vasculaire chronique et récurrente. La thrombose dans la microcirculation entraîne une diminution de la perfusion, suivie d’une ulcération de la peau. Les ulcérations touchent exclusivement le membre inférieur, en particulier la région malléolaire. Le guide S1 sur le diagnostic et le traitement de la livedovasculopathie, qui sera publié en 2021, indique que la confirmation histologique du diagnostic ne peut être obtenue qu’au stade aigu de la maladie, au stade de l’ischémie [3]. Il est recommandé de prélever un échantillon suffisamment grand (si possible une biopsie du fuseau) de la zone périphérique de la zone affectée. Elle se caractérise par des dépôts de fibrine souvent difficiles à détecter dans les parois des vaisseaux, ainsi que par des thrombus de fibrine, qui se trouvent principalement dans les vaisseaux du derme supérieur et moyen. Plusieurs paramètres procoagulants sont décrits dans la livedovasculopathie. Il s’agit notamment de défauts d’activation du plasminogène endothélial, de dysfonctionnements plaquettaires et d’une augmentation de la production de fibrine. Les dépôts de fibrine et la formation de thrombus entraînent une ischémie des tissus et, par la suite, des ulcérations. Une atrophie blanche peut se former comme manifestation chronique et point final des processus de remodelage cicatriciel. Il s’agit d’une cicatrice en forme d’éclair ou d’étoile, semblable à de la porcelaine.
Afin d’éviter la progression de l’infarctus cutané chronique récurrent avec transformation cicatricielle du site de manifestation, il est nécessaire de commencer un traitement médicamenteux à un stade précoce. La stratégie de traitement a changé par rapport au passé. “Aujourd’hui, les patients sont traités par rhéologie, c’est-à-dire par héparine de bas poids moléculaire”, explique le conférencier. En l’espace de quelques jours, les patients ressentent nettement moins de douleurs. Les DOAK (anticoagulants oraux directs) constituent une autre option thérapeutique. Selon le professeur Dissemond, on peut tout au plus ajouter de la cortisone pendant les premiers jours.
La calciphylaxie – un danger de mort en l’absence de traitement
Dans 90 à 95% des cas, cette vasculopathie, également appelée “artériolopathie urémique calcifiante”, concerne des patients atteints d’insuffisance rénale terminale. Le site de prédilection est le membre inférieur, les manifestations typiques sont la nécrose et l’érythème livide. Le stade initial se caractérise par une induration douloureuse de la peau qui peut faire penser à une névralgie zostérienne [4]. La peau présente souvent une coloration livide rougeâtre, avec des motifs réticulaires. Le développement d’une palpation en forme de cuir et de plaque est typique. Le tableau complet se caractérise par des ulcérations profondes qui dépassent nettement le derme. Le plus souvent, la bordure présente une texture irrégulière, comme une carte. Il s’agit d’une pathologie grave. Sur le plan thérapeutique, il peut être utile d’essayer un traitement au thiosulfate de sodium. Dans deux séries de cas non contrôlés de patients atteints de calciphylaxie (n=199), le thiosulfate de sodium a été utilisé dans le cadre d’une approche thérapeutique multimodale [5,6]. Une dose de 25 g de thiosulfate de sodium a été administrée sous forme de perfusion à la fin/vers la fin de chaque hémodialyse. La durée du traitement peut être de plusieurs semaines ou de plusieurs mois [4].
Syndrome de Klinefelter – risque accru de phlébothrombose
Il existe également des causes génétiques de troubles graves de la cicatrisation. Une analyse génétique est nécessaire pour le diagnostic. Le syndrome de Klinefelter se caractérise par une aberration chromosomique numérique sous la forme d’une trisomie (47, XXY). L’incidence des phlébothromboses est jusqu’à 20 fois plus élevée que dans la population normale en raison de divers facteurs thrombogènes. Les plaies chroniques correspondent généralement à un ulcère post-thrombotique.
Congrès : Austrian Wound Association
Littérature :
- Dissemond J : Causes rares de plaies chroniques (“maladies orphelines”). Dr. med. Joachim Dissemond, Austrian Wound Association, 25.03.2022
- Orphanet, www.orpha.net (dernière consultation 17.08.2022)
- Görge T, et al. : S1-Leitlinie Diagnostik und Therapie der Livedovaskulopathie, 2021 ; 013-098.
- Brandenburg VM, et al : Calciphylaxie. Dtsch Med Wochenschr 2015 ; 140 : 347-351.
- Nigwekar SU, et al : Thérapie au thiosulfate de sodium pour l’artériopathie urémique calcique. Clin J Am Soc Nephrol 2013 ; 8 : 1162-1170.
- Zitt E, et al : Use of sodium thiosulphate in a multi -interventional setting for the treatment of calciphylaxis in dialysis patients. Nephrol Dial Transplant 2013 ; 28 : 1232-1240.
- Hobbs MM, Ortega-Loayza AG : Pyoderma gangrenosum : From historical perspectives to emerging investigations. Int Wound J 2020 ; 17(5) : 1255-1265.
DERMATOLOGIE PRATIQUE 2022 ; 32(4) : 40-41
PRATIQUE DU MÉDECIN DE FAMILLE 2022 ; 17(9) : 16-17