
La sarcoïdose peut survenir à tout âge et affecte typiquement les poumons, mais plusieurs autres organes peuvent également être impliqués. L’établissement du diagnostic se fait en plusieurs étapes, il n’existe pas de tests sanguins spécifiques permettant de confirmer ou d’exclure la sarcoïdose. Histologiquement, la sarcoïdose se caractérise par des granulomes épithélioïdes non nécrosants. Une fois qu’une sarcoïdose pulmonaire a été identifiée, il convient de rechercher des atteintes extrathoraciques pertinentes, en particulier les manifestations cardiaques qui peuvent mettre la vie en danger.
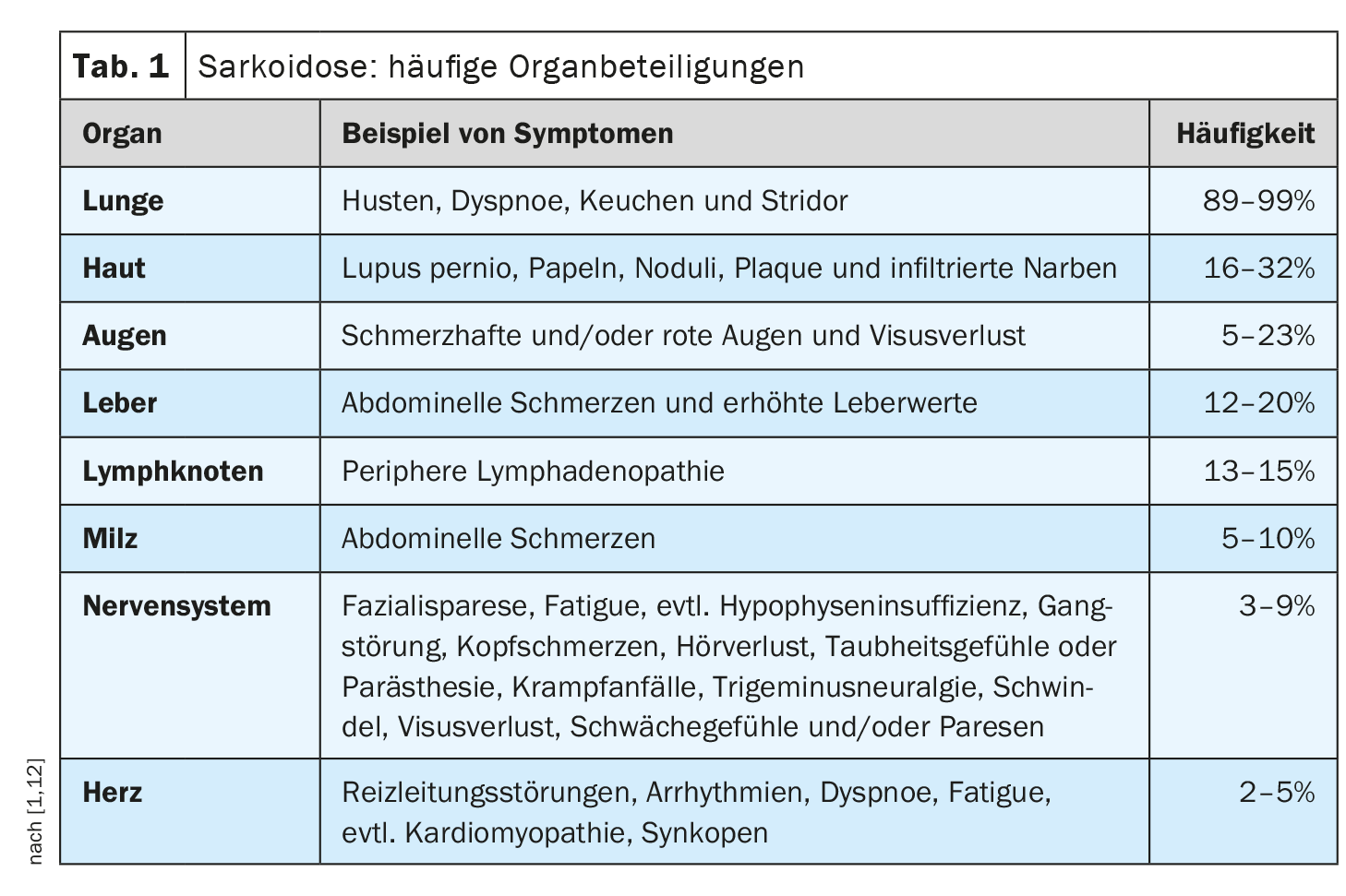
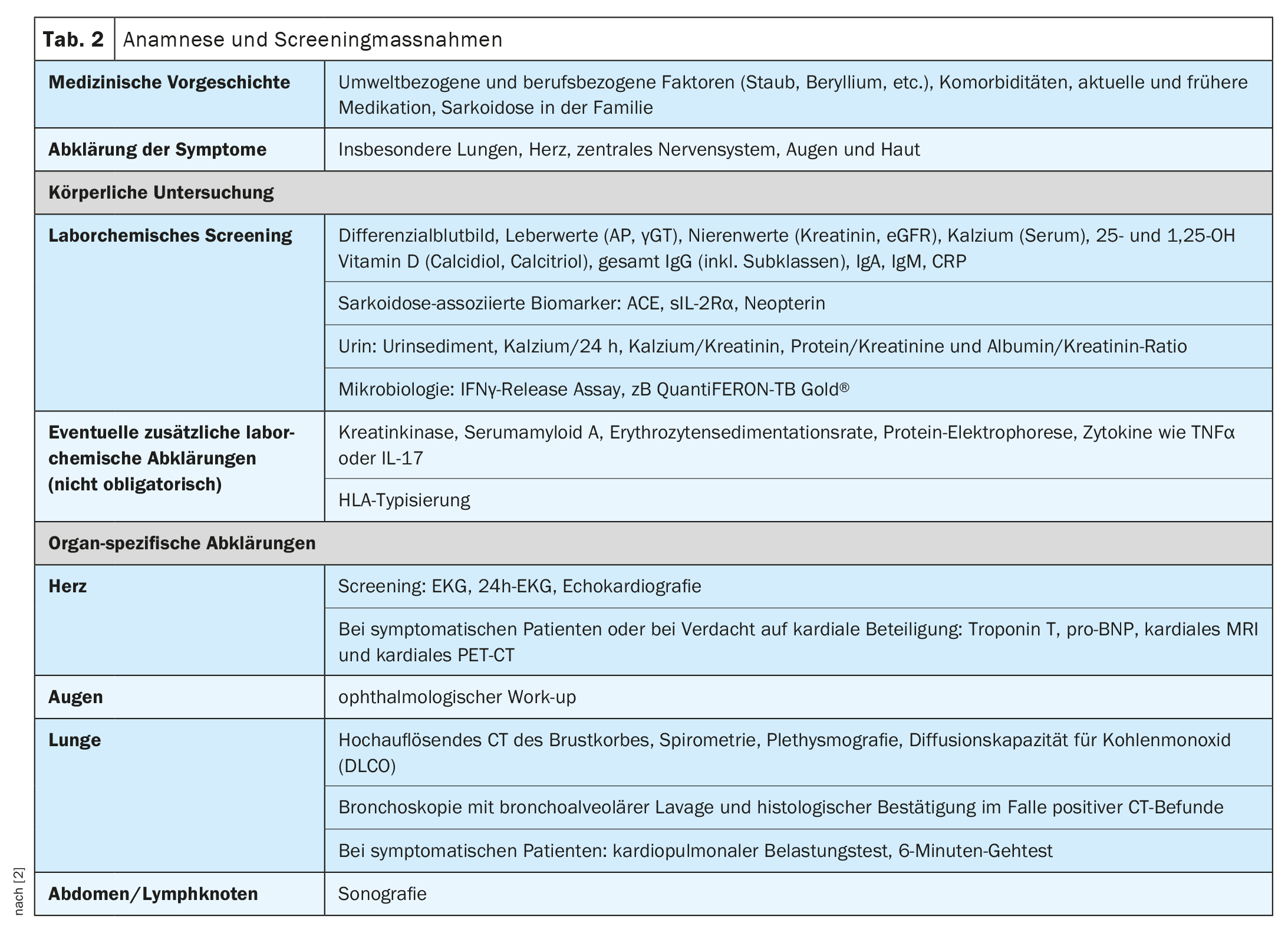
L’évolution clinique et le pronostic de la sarcoïdose sont très hétérogènes et dépendent de l’organe atteint, a expliqué le professeur Jörg D. Seebach, médecin chef de Service d’immunologie et allergologie, Hôpitaux universitaires de Genève [1]. Les poumons, les ganglions lymphatiques, la peau et les yeux sont le plus souvent touchés, tandis que les manifestations cardiaques, rénales et neurologiques sont moins fréquentes mais s’accompagnent d’une morbidité plus élevée [2]. Le tableau 1 donne un aperçu des manifestations organiques les plus fréquentes. Le diagnostic de la sarcoïdose repose principalement sur les caractéristiques cliniques et radiologiques, l’identification de granulomes non nécrosants dans un ou plusieurs échantillons de tissus, et l’exclusion des autres causes de la maladie granulomateuse [1,3]. Parfois, la sarcoïdose évolue de manière autolimitée – environ la moitié des patients atteints de sarcoïdose connaissent une rémission spontanée dans les deux ans et beaucoup d’autres dans les cinq ans suivant le début des symptômes [4]. Mais il existe aussi des formes d’évolution plus graves. Les causes de décès les plus fréquentes chez les patients atteints de sarcoïdose sont l’insuffisance respiratoire en cas de fibrose pulmonaire avancée et l’atteinte cardiaque ou neurologique sévère [3]. Le tableau 2 résume les recommandations concernant l’anamnèse et les mesures de dépistage et le tableau 3 les investigations en cas de suspicion d’atteinte de certains organes.
Les procédures bronchoscopiques sont peu invasives
Si une sarcoïdose est suspectée sur la base des symptômes cliniques et des résultats radiologiques, le conférencier recommande de demander une biopsie [1]. En principe, les échantillons histologiques doivent être prélevés à l’endroit le moins invasif et offrant les meilleures possibilités de diagnostic. Comme les poumons sont concernés dans la plupart des cas, la bronchoscopie est une procédure sûre et peu invasive [2]. Il existe plusieurs procédures de diagnostic bronchoscopique, y compris la biopsie endobronchique (muqueuse), la biopsie pulmonaire transbronchique (TBLB), ou l’aspiration transbronchique à l’aiguille (TBNA) des ganglions lymphatiques hilaires/médiastinaux et le lavage broncho-alvéolaire (LBA). Des échantillons biologiques extrapulmonaires, par exemple de la peau, des glandes parotides ou lacrymales, des ganglions lymphatiques palpables ou des lésions conjonctivales, sont également possibles, mais moins spécifiques. Dans tous les cas, l’histologie doit être associée à des manifestations cliniques et radiologiques compatibles et à l’exclusion d’autres maladies. En présence d’une confirmation histologique sur des sites extrapulmonaires et d’une atteinte pulmonaire concomitante avec suspicion d’infection, telle qu’une pneumopathie cavitaire, une bronchoscopie peut être nécessaire pour exclure les causes infectieuses telles que les mycobactéries et les champignons [2].
La manifestation pulmonaire est l’organe le plus fréquemment impliqué
L’atteinte des poumons et/ou des ganglions lymphatiques médiastinaux/hilaires est l’atteinte d’organe la plus fréquente, qui se produit chez environ 80 à 90% des patients atteints de sarcoïdose. Une manifestation pulmonaire est associée à une participation parenchymateuse et à des granulomes périvasculaires. Les symptômes les plus courants sont une sensation de pression dans la poitrine, une toux sèche et une dyspnée. Par la suite, une fibrose peut se développer et entraîner une insuffisance respiratoire [5]. La sarcoïdose pulmonaire pouvant être associée à des schémas obstructifs, restrictifs, mixtes ou normaux, les résultats des explorations fonctionnelles respiratoires sont très peu spécifiques, mais importants pour l’évaluation de la sévérité, l’indication du traitement et la réponse au traitement. La pneumopathie interstitielle (PID) est une manifestation typique aux stades 2, 3 et 4, allant de manifestations infracliniques à la fibrose pulmonaire au stade terminal (stade 4). Cette dernière est une lésion irréversible de l’organe, tandis que la DLI légère à modérée due à la sarcoïdose est une symptomatologie qui peut être traitée.

Les manifestations cardiaques sont plus rares, mais leur pronostic est défavorable
Alors que près de 90% des patients atteints de sarcoïdose présentent une atteinte pulmonaire, la survenue d’une sarcoïdose cardiaque est plutôt rare, avec une prévalence de 2 à 7% [6]. Toutefois, chez les patients présentant une sarcoïdose extracardiaque confirmée, il est recommandé de rechercher une implication cardiaque au moyen d’un ECG [7]. En effet, la sarcoïdose cardiaque est une manifestation qui peut potentiellement mettre la vie en danger [2]. Les techniques d’imagerie modernes peuvent être utiles pour une détection précoce. Il s’agit notamment d’une IRM cardiaque avec la technique d’amélioration du gadolinium tardif (LGE) et de la TEP-FDG [8]. L’implication cardiaque peut se manifester par des arythmies ventriculaires, un bloc cardiaque de haut niveau ou une insuffisance cardiaque chronique due à une infiltration granulomateuse myocardique et/ou, aux stades ultérieurs de la maladie, par une fibrose [2]. Les symptômes possibles sont des douleurs au niveau de la cage thoracique, des palpitations, des vertiges et des syncopes. Jusqu’à 25% d’entre eux sont victimes d’une mort subite d’origine cardiaque. En résumé, un diagnostic et un traitement précoces de l’atteinte cardiaque sont essentiels [2,9]. Les manifestations typiques de la sarcoïdose cardiaque sont les troubles de la conduction, les arythmies ventriculaires et l’insuffisance cardiaque [13]. Dans environ un quart des cas, la sarcoïdose cardiaque apparaît de manière isolée, sans implication pulmonaire, selon le professeur Seebach [1]. Ceci est souvent associé à un moins bon pronostic par rapport à la sarcoïdose systémique avec atteinte cardiaque. Les diagnostics différentiels importants sont : la myocardite lymphocytaire, certaines cardiomyopathies génétiques, l’augmentation physiologique de l’absorption du FDG en cas d’insuffisance cardiaque [1].
Diagnostic différentiel : que faut-il prendre en compte ?
“Le diagnostic différentiel est très important”, a souligné le conférencier [1]. L’analyse histopathologique est très importante. La sarcoïdose est caractérisée par des granulomes compacts, non nécrosants, avec une distribution périlymphatique le long des faisceaux bronchovasculaires, paraseptaux et pleuraux [2]. Dans les biopsies pulmonaires ouvertes, la sarcoïdose est souvent associée à une vascularite granulomateuse sans destruction des parois vasculaires. Au cours de la progression de la maladie, la fibrose hyalinisée domine avec des restes de granulomes. La bérylliose chronique est un diagnostic différentiel essentiel de la sarcoïdose, en particulier chez les patients exposés aux métaux. La maladie granulomateuse induite par l’infliximab doit également être prise en considération. Ces entités se distinguent bien de la sarcoïdose sur le plan histologique. Les autres diagnostics différentiels comprennent les malignités (lymphomes, carcinomes), les collagénoses vasculaires (lupus érythémateux disséminé, syndrome de Sjögren, cirrhose biliaire primitive, arthrite granulomateuse familiale), les infections (VIH, tuberculose), vascularite (granulomatose avec polyangéite, artérite de Takayasu, artérite à cellules géantes), pneumonie d’hypersensibilité, pneumoconiose/pneumoconiose, maladie liée aux IgG4 et diverses maladies d’immunodéficience) [2,10,11]. La morphologie et les schémas de distribution des granulomes concernés sont différents de ceux de la sarcoïdose.
Les maladies infectieuses (tuberculose, Myobacterium avium, histoplasmose, coccidiomycose et maladie de Whipple) présentent une distribution péribronchique ou aléatoire des granulomes et sont souvent associées à une nécrose. L’utilisation de colorations spéciales (Ziehl-Neelsen, auramine et coloration à l’argent) ou de tests PCR pour le complexe Mycobacterium tuberculosis et les mycobactéries atypiques peut être révélatrice pour la détection de l’agent pathogène. Contrairement à la sarcoïdose, la pneumonie d’hypersensibilité se caractérise par des amas lâches d’histiocytes à proximité des bronchioles. La granulomatose avec polyangéite (anciennement appelée granulomatose de Wegener) se caractérise par une nécrose basophile entourée d’un infiltrat cellulaire contenant des cellules géantes. La granulomatose nodulaire sarcoïdienne est parfois considérée comme une variante de la sarcoïdose et se caractérise également par une nécrose extensive qui, contrairement à la granulomatose avec polyangéite, est éosinophile et marquée par de nombreux granulomes compacts accompagnés d’une vascularite granulomateuse sans destruction des parois vasculaires.
Les diagnostics différentiels en cas de manifestations neurologiques doivent être basés sur les résultats de l’IRM (en particulier les lésions périventriculaires, focales vs les lésions parenchymateuses ou méningées). Les nouvelles techniques d’IRM ont une sensibilité optimisée, mais en raison d’un manque de spécificité, la délimitation du diagnostic reste un défi. [2]. L’éventail des diagnostics différentiels s’étend des maladies auto-immunes, inflammatoires ou idiopathiques (par ex. sclérose en plaques, maladie du spectre de la neuromyélite optique, LED, syndrome de Sjögren, maladie de Behcet, vascularite primaire du SNC) aux entités infectieuses (par ex. tuberculose, maladie de Lyme, neurosyphilis, taxoplasmose) et aux néoplasmes (néoplasmes primaires du SNC, lymphomes et autres) [2].
Littérature :
- «Sarcoidosis: Beyond the Lungs and Lymph Nodes», Prof. Dr. med. Jörg D. Seebach, Allergy and Immunology Update, Grindelwald, 27.–29.1.2023.
- Franzen DP, et al.: Sarcoidosis – a multisystem disease. Swiss Med Wkly. 2022;152: w 30049.
- Graf L, Geiser T: Das Chamäleon unter den Systemerkrankungen: «Die Sarkoidose». Swiss Med Forum 2018; 18(35): 695–701.
- Valeyre D, et al.: Sarcoidosis. Lancet (London, England) 2014; 383(9923): 1155–1167.
- Lichtenberger N: Diagnostik und Therapie der Kardialen und pulmonalen Sarkoidose. https://opus.bibliothek.uni-wuerzburg.de, (dernière consultation 23.02.2023)
- Costabel U, et al. [Cardiac sarcoidosis: diagnostic and therapeutic algorithms]. Pneumologie (Stuttgart, Germany) 2014; 68(2): 124–132.
- Birnie DH, et al.: HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart rhythm 2014; 11(7): 1305–1323.
- Giblin GT, et al.: Cardiac Sarcoidosis: When and How to Treat Inflammation. Card Fail Rev 2021;7:e17. doi: 10.15420/cfr.2021.16.
- Hamzeh N, et al.: Pathophysiology and clinical management of cardiac sarcoidosis. Nat Rev Cardiol 2015; 12(5): 278–288.
- Valeyre D, et al.: Clinical presentation of sarcoidosis and diagnostic work-up. Semin Respir Crit Care Med 2014; 35(3): 336–351.
- Spagnolo P, et al.: Pulmonary sarcoidosis. Lancet Respir Med 2018; 6(5): 389–402.
- Grunewald J, et al.: Sarcoidosis. Nat Rev Dis Primers 2019; 5(1): 45.
- Birnie D, et al.: Cardiac Sarcoidosis. Clinics in chest medicine 2015; 36(4): 657–668.
HAUSARZT PRAXIS 2023; 18(5): 40–42
Photo de couverture : Hellerhoff, wikimedia









