Les nodules thyroïdiens sont un phénomène bien connu, les carcinomes médullaires de la thyroïde ne le sont heureusement pas. Il s’agit d’une dégénérescence maligne des cellules C de la thyroïde, qui sont parafolliculaires et ne peuvent pas stocker l’iode. Comme elle sécrète de la calcitonine, celle-ci peut être utilisée comme marqueur tumoral.
Les nodules thyroïdiens sont souvent diagnostiqués dans la pratique, mais heureusement rarement un carcinome médullaire de la thyroïde. Seulement 3% environ de tous les cancers de la thyroïde concernent le carcinome médullaire de la thyroïde (MTC), qui présente certaines particularités : Il s’agit d’une dégénérescence maligne des cellules C de la thyroïde, qui sont parafolliculaires et ne peuvent pas stocker l’iode ; il sécrète de la calcitonine (Ctn) et de l’ACE, qui sont utilisés comme marqueurs tumoraux ; un quart des cas sont familiaux, dans le cadre de la néoplasie endocrinienne multiple de type 2 (MEN2). La seule option pour une éventuelle guérison de la MTC est un diagnostic précoce et une chirurgie adéquate. Ceci est rendu possible par l’utilisation systématique du dosage du Ctn lors de l’examen d’un goitre noueux et, dans le cas de la variante familiale, de l’analyse génétique moléculaire du protooncogène RET dans le cadre du dépistage familial. Même chez les personnes non guéries, le CTM a un pronostic relativement favorable en raison de sa croissance lente et d’une qualité de vie relativement bonne. Une stratégie de suivi “surveillance active” adaptée au risque est possible dans de nombreux cas. Un CTM au stade de métastases à distance symptomatiques et progressives peut aujourd’hui être traité par des inhibiteurs de tyrosine kinase (ITK) [1,2].
Clinique et diagnostic du carcinome médullaire de la thyroïde (CMT)
Cliniquement, le CTM médullaire ne diffère pas beaucoup des autres cancers de la thyroïde : nodule croissant dans la thyroïde, gêne non spécifique au niveau du cou, développement d’un gonflement des ganglions lymphatiques cervicaux, et au stade métastatique avancé, des diarrhées marquées induites par la tumeur peuvent apparaître. Aujourd’hui, la MTC est diagnostiquée soit de manière fortuite lors de l’histologie d’une pièce opératoire thyroïdienne, soit lors d’un examen préopératoire dans le cadre de l’évaluation d’un nodule thyroïdien. (Fig.1). Les éléments d’orientation sont un nodule suspect à l’échographie de la thyroïde (écho faible, microcalcification, bords flous, extension plus profonde que large), des ganglions lymphatiques cervicaux suspects, une ponction à l’aiguille fine suspecte et un taux de Ctn élevé. Un taux de Ctn supérieur à 100 pg/ml est le résultat le plus sensible et le plus spécifique. Les patients concernés sont ceux en 4. et 5e décennie de vie. Chez les patients plus jeunes, il faut penser à la variante familiale (MEN2), qui peut parfois se manifester par le biais de maladies concomitantes telles que le phéochromocytome et l’hyperparathyroïdie primaire. Dans le cadre de l’examen d’une augmentation de l’ACE, il faut également penser à la MTC, qui est rare.
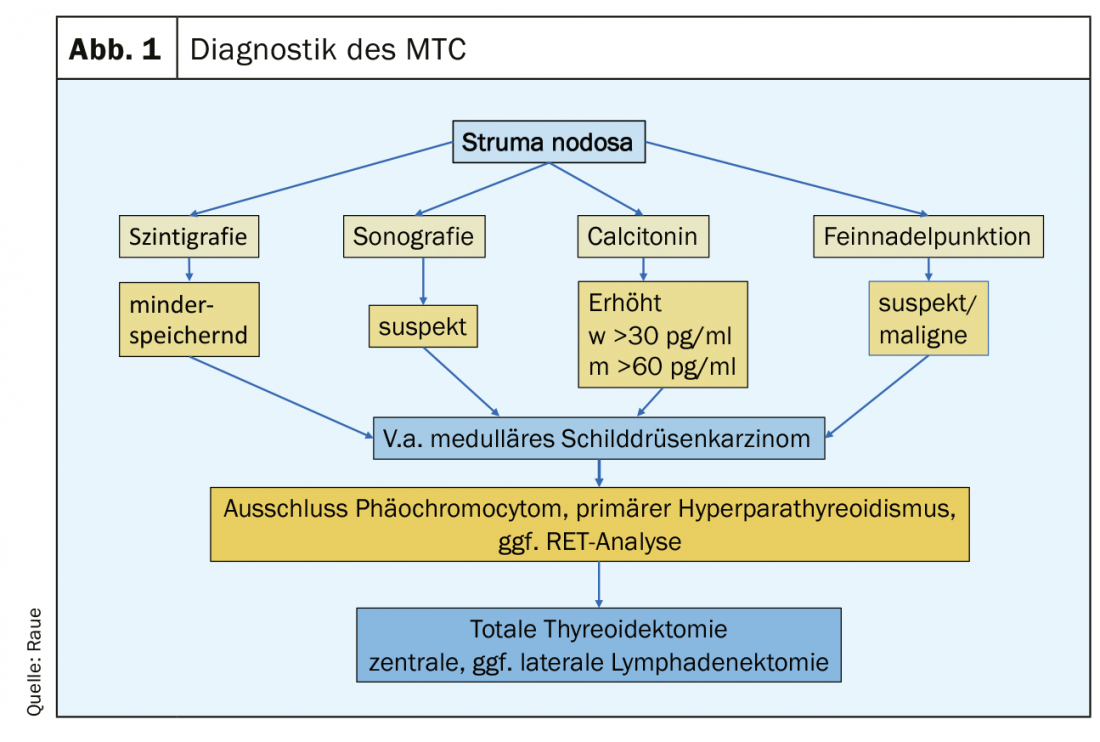
Dépistage de la calcitonine dans le cadre de l’évaluation du goitre noueux
Ctn est corrélé à la masse tumorale et constitue un marqueur tumoral sensible et spécifique pour la détection précoce et le suivi d’une MTC. La détermination de routine du Ctns lors de l’examen du goitre noueux permet un diagnostic précoce [3]. Des taux légèrement élevés de Ctn sont observés en cas d’hyperplasie des cellules C, qui peut être un précurseur d’une MTC héréditaire, mais qui est également observée comme un phénomène concomitant “bénin” dans d’autres maladies thyroïdiennes [4]. Avec le développement de nouveaux tests Ctn sensibles et spécifiques entièrement automatisés (tests immunologiques en chimioluminescence avec une sensibilité de 0,5 pg/ml et valeurs de référence supérieures spécifiques au sexe) [5,6], les taux basaux de Ctn pour le dépistage du goitre noueux sont devenus plus importants.
Les taux de Ctn préopératoires supérieurs à 100 pg/ml indiquent une MTC dans près de 100% des cas, tandis que les taux de Ctn compris entre 10-20 pg/ml ont un taux de détection de MTC inférieur à 5% [7]. En tenant compte de la sensibilité et de la spécificité, les valeurs de cut-off du Ctns basal en fonction du sexe pour la recommandation d’opérer une MTC présumée sont d’environ 30 pg/ml pour les femmes et d’environ 60 pg/ml pour les hommes. La zone grise de Ctn est de 20-30 pg/ml pour les femmes, et de 30-60 pg/ml pour les hommes, dans laquelle 6-13% des petits MTC ont été manqués [8]. Des niveaux croissants de Ctn sont plus susceptibles d’indiquer une MTC, auquel cas la chirurgie devrait être recommandée. Les mesures chirurgicales permettent de guérir un MTC dans presque 100% des cas jusqu’à un taux de Ctn de 100 pg/ml [9].
Proto-oncogène RET – Mutations germinales dans le MEN2
Chez environ 25% des patients atteints de MTC, une mutation germinale dans le proto-oncogène RET peut être détectée comme indication d’un syndrome MEN2. Cliniquement, on distingue le MEN2A, avec le développement possible d’un phéochromocytome et d’une hyperparathyroïdie, du MEN2B, qui présente un habitus typique, une ganglioneuromatose des muqueuses et le développement possible d’un phéochromocytome. Les antécédents familiaux ne permettent pas d’identifier toutes les familles, car les familles sont souvent petites ou il s’agit d’une hMTC à manifestation tardive et à faible pénétrance. Dans un faible pourcentage, on observe une mutation “de novo” chez le patient, les autres membres de la famille ne sont pas touchés. Dans une grande série, on a trouvé une mutation germinale dans le proto-oncogène RET chez 12% des MTC apparemment sporadiques. C’est pourquoi tous les patients atteints de MTC doivent faire l’objet d’un examen de génétique moléculaire pour détecter des mutations germinales dans le gène RET.
Dans la forme héréditaire du MTC, les mutations causales sont caractérisées dans presque toutes les familles, généralement des mutations ponctuelles dans 8 exons différents du proto-oncogène RET. La détection de ces mutations par la génétique moléculaire, associée à la détermination du Ctn dans les familles, permet de réaliser une thyroïdectomie précoce chez les enfants et les adolescents atteints, avec guérison de la MTC. Le moment optimal de la thyroïdectomie chez les porteurs du gène d’une mutation RET est aujourd’hui recommandé sur la base de la classification du risque de la mutation RET spécifique en risque modéré, élevé et très élevé de développement précoce du CTM (pénétrance précoce ou tardive) (tableau 1) [1]. Le niveau de Ctn définit individuellement la thyroïdectomie prophylactique à planifier précocement, de sorte que dans le meilleur des cas, une lymphadénectomie supplémentaire ne soit pas nécessaire.

Jusqu’à 50% des patients MEN2 développent des phéochromocytomes au cours de leur vie, en fonction de leur génotype, généralement après la manifestation d’une MTC. Les phéochromocytomes peuvent être multifocaux et bilatéraux [10]. Les patients présentant des mutations RET-634 et -918 sont particulièrement touchés, plus rarement ceux présentant des mutations dans les exons 13-15. Jusqu’à 10% des patients MEN2 peuvent développer une hyperparathyroïdie primaire en fonction du génotype, les patients porteurs d’une mutation RET-918 ne sont pas concernés. D’autres manifestations extrathyroïdiennes, telles que la ganglioneuromatose dans le MEN2B ou le lichen amyloïde interscapulaire, dépendent également du génotype.
Dans le tissu tumoral des MTC sporadiques, une mutation somatique du gène RET, généralement RET-M918T, est souvent détectée, curieusement moins souvent dans les petites tumeurs, plus souvent dans les grandes et le plus souvent dans les filiations ganglionnaires et les métastases à distance, ce qui suggère que cette mutation ne constitue pas l’événement primaire dans la tumorigenèse. Dans les tissus tumoraux où l’on ne trouve pas de mutation RET, on parvient souvent à détecter une mutation RAS. La mise en évidence de la mutation somatique RET a une importance pronostique et constitue une condition préalable au traitement par les inhibiteurs sélectifs de la tyrosine kinase RET (LOXO-292, BLU-667) qui seront probablement bientôt disponibles [11].
Opération du MTC
Le traitement primaire de la MTC est chirurgical. La thyroïdectomie totale est l’intervention minimale, complétée par une dissection centrale, éventuellement latérale, unilatérale/bilatérale des ganglions lymphatiques (LK), en fonction du Ctn mesuré en préopératoire et du stade de la tumeur. Dans le cas d’une MTC découverte par hasard à l’histologie, le niveau de Ctn postopératoire devrait décider de la procédure à suivre ; si le Ctn est faible et non mesurable, aucune autre opération n’est nécessaire ; si le Ctn est élevé et en fonction du stade de la tumeur, une opération complète (TX totale et dissection des LK centraux et latéraux) devrait être réalisée. Un traitement chirurgical curatif n’est possible que si aucune métastase à distance, aucune infiltration dans les parties molles n’a été décrite en histologie primaire et si moins de 5 à 10 LK cervicaux atteints sont détectables dans l’histologie précédente ou moins de 3 compartiments atteints après lymphadénectomie systématique [12]. Si cette situation favorable existe, il convient de tenter un traitement chirurgical curatif. En cas de MTC héréditaire, une thyroïdectomie totale est toujours indiquée, car toute cellule C peut en principe se développer en MTC. Parmi les MTC diagnostiqués via un nodule thyroïdien palpable, 70% ont déjà des métastases dans les ganglions lymphatiques cervicaux et 13% ont des métastases à distance dans le foie, les poumons ou les os [13]. La survie à 10 ans est d’environ 61-81% [14], en fonction du stade de la tumeur au moment du diagnostic, de l’âge, du sexe, des taux de Ctn pré- et postopératoires. Au cours de la dernière décennie, en raison d’un diagnostic de plus en plus précoce, de stades tumoraux plus favorables et de stratégies chirurgicales améliorées, la survie a augmenté globalement, mais aussi aux stades tumoraux plus avancés [14,15].
Chez les patients atteints de MEN2 détectés lors du dépistage familial, le moment de la “thyroïdectomie prophylactique” est déterminé par la mutation RET et le taux de Ctn (cf. Tab. 1). Un diagnostic précoce et une chirurgie complète ont permis d’augmenter la survie à 5 ans spécifique à la maladie de tous les MTC à 89% [14].
Suivi du MTC
En postopératoire, les résultats suivants doivent être disponibles pour planifier un suivi structuré : l’histologie, éventuellement l’immunohistologie Ctn de la tumeur, la classification selon le schéma pTNM, le résultat de l’analyse RET pour la classification en variante héréditaire ou sporadique et le taux de Ctn postopératoire. On peut définir 3 groupes de patients à risque en ce qui concerne la réponse au traitement primaire et le pronostic : excellent (Ctn non mesurable faible), biochimiquement incomplet (Ctn mesurable, mais pas de tissu tumoral détectable à l’imagerie) et structurellement incomplet (Ctn nettement élevé, détection de ganglions lymphatiques atteints de métastases ou de métastases à distance). (Fig. 2). Le calcul du temps de doublement des marqueurs tumoraux (Ctn/CEA) et l’imagerie sont ensuite effectués pour détecter la croissance possible de la tumeur, puis le groupe de risque est adapté. [16,17]. Les intervalles de suivi sont adaptés au risque et sont effectués tous les 3 mois à une fois par an, en fonction de la taille et de l’emplacement de la tumeur résiduelle/des métastases et du degré de progression de la tumeur [18]. En fonction de la localisation des métastases suspectées, les techniques d’imagerie à envisager sont l’échographie du cou et de l’abdomen, la tomodensitométrie avec produit de contraste, l’IRM, la scintigraphie osseuse et, le cas échéant, la TEP-FDG ou la TEP-F-DOPA (Fig. 1).
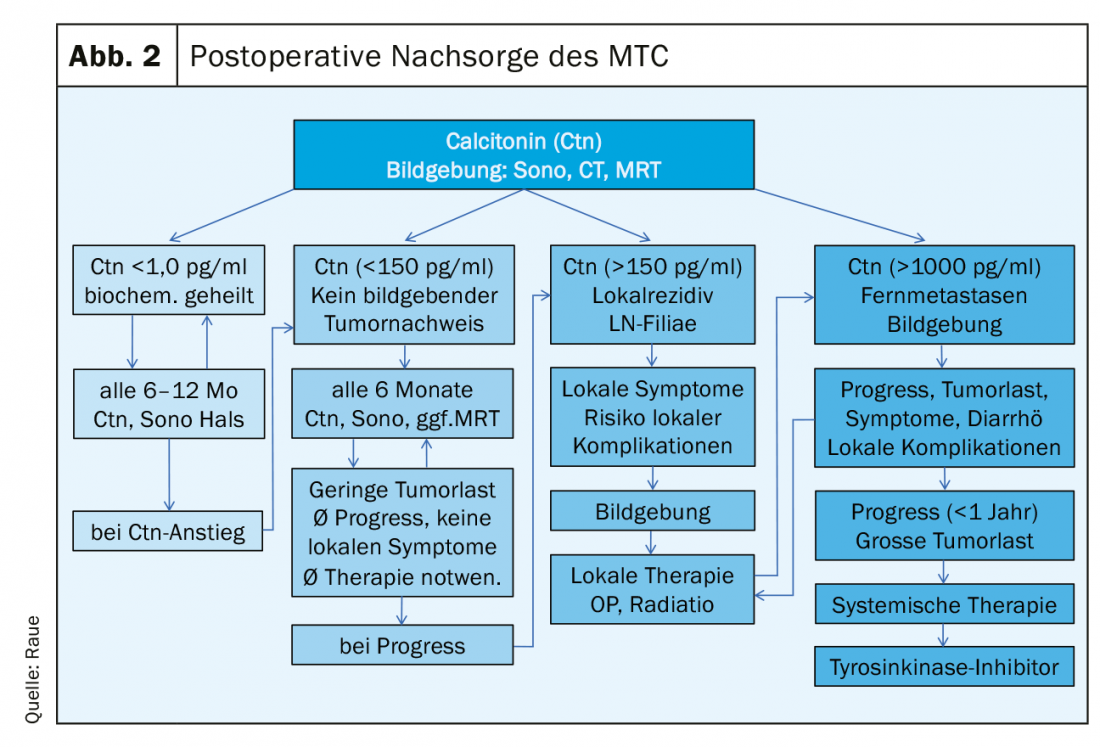
Groupes à risque dans le cadre du suivi
En cas d’excellente réponse, le taux de Ctn n’est pas bas de manière mesurable : on peut alors supposer une guérison biochimique du patient, à condition que l’histologie soit correcte (immunohistologie Ctn obligatoire). Pendant les 2 premières années, il suffit d’effectuer un suivi semestriel avec échographie du cou, dosage de la Ctn et de l’ACE, vérification du traitement substitutif par thyroxine (objectif : TSH dans la norme), voire administration de calcium et de calcitriol en cas d’hypoparathyroïdie postopératoire (objectif : calcium sérique dans la limite inférieure 2,0-2,3 mmol/l) [19]. Ensuite, s’il n’y a pas d’augmentation du Ctn, on peut passer à des contrôles annuels. Le pronostic est excellent et ne diffère pas de celui de la population normale.
En cas de réponse biochimique incomplète, le taux de Ctn est généralement légèrement à modérément élevé de manière persistante (généralement inférieur à 1000 pg/ml). On peut supposer qu’il reste du tissu tumoral. Si l’opération précédente n’était pas adéquate, une opération complémentaire adéquate est effectuée après un “staging”. Cependant, si ces limites (voir la section Opération de la MTC) sont dépassées, une approche curative n’est plus possible, c’est pourquoi toutes les autres mesures thérapeutiques doivent être évaluées de manière critique en tenant compte du risque de morbidité et de la qualité de vie généralement bonne. Souvent, malgré une recherche intensive, on ne trouve pas de corrélat tumoral certain lorsque la Ctn n’est que modérément élevée, jusqu’à 150 pg/ml. La progression de la maladie tumorale au cours de l’évolution peut être très variable et peut être relativement bien évaluée sur la base du temps de doublement du Ctn et du CEA [20]. La condition préalable à une bonne déclaration est d’avoir au moins 4 valeurs Ctn sur 2 ans. Les patients dont le temps de doublement des marqueurs tumoraux était inférieur à 24 mois présentaient également une progression sur le plan de la morphologie de l’image dans 94% des cas. Lorsque les temps de doublement des marqueurs tumoraux s’étendaient sur 24 mois, 86% des patients n’avaient pas de croissance tumorale détectable [21]. Par la suite, des contrôles semestriels de l’évolution sont généralement suffisants. La fréquence des examens d’imagerie peut être planifiée en fonction du résultat primaire, de la croissance au cours de l’évolution (critères RECIST) et du temps de doublement des marqueurs tumoraux. Un diagnostic maximal rapproché sans conséquence thérapeutique n’est pas utile, il suffit souvent par exemple d’une échographie tous les 6 mois et d’un scanner/IRM tous les 12 mois.
En cas de réponse structurellement incomplète, le taux de Ctn est nettement élevé (généralement plus de 1000 pg/ml) : Dans ce cas, on peut supposer qu’il s’agit d’un tissu tumoral infiltrant localement, de métastases dans les ganglions lymphatiques cervicaux ou d’une métastase à distance, généralement dans les poumons, le foie et/ou les os. Une approche curative n’est plus possible, les mesures palliatives sont au premier plan [22].
Les ré-opérations d’une récidive loco-régionale sous approche palliative sont particulièrement utiles en cas de récidive locale progressive ou de filiation ganglionnaire douloureuse dans la région centrale du cou (proximité/infiltration de la trachée ou de l’œsophage) afin de réduire les complications locales. Il n’est pas judicieux de soumettre toute nouvelle métastase détectable au niveau cervical à une opération immédiate, car cela conduit à de nombreuses opérations qui n’aboutissent pas. Il n’y a aucune preuve que ces opérations aient un effet positif sur l’évolution globale, elles sont souvent associées à des effets secondaires tels que la parésie récurrentielle, l’hypoparathyroïdie et la paralysie des muscles du bras. La radiothérapie est utile en cas de récidives locales ou médiastinales progressives et inopérables. Comme il est plus difficile d’opérer après une radiothérapie, il faut toujours commencer par évaluer l’opérabilité d’un point de vue palliatif.
Les métastases hépatiques sont fréquentes, provoquent généralement peu de symptômes et sont rarement une indication de chirurgie. Les métastases hépatiques nettement progressives et douloureuses doivent être traitées. Le plus souvent, il s’agit de métastases multiples et diffuses. Le traitement local par (chimio)embolisation ou radiothérapie interne sélective (SIRT) est d’une efficacité limitée. Un traitement systémique par des inhibiteurs de tyrosine kinase est indiqué en cas de progression rapide.
Les métastases osseuses sont plus rarement ostéolytiques, fracturaires ou douloureuses. En cas de métastases osseuses douloureuses, une radiothérapie externe est indiquée, et en cas de risque de fracture, une radiothérapie externe ou une intervention chirurgicale. Un traitement par bisphosphonate/denosumab peut être ajouté individuellement, surtout en cas de douleurs ou de risque de fracture, en particulier en cas de métastases ostéolytiques [23].
La chimiothérapie est peu efficace dans le cas du MTC métastasé. Depuis une dizaine d’années, les inhibiteurs de tyrosine kinase font l’objet d’études dans le cadre du traitement du MTC métastatique avancé. Depuis mai 2012, le vandétanib, un inhibiteur de tyrosine kinase, est disponible en Suisse pour le traitement du MTC agressif et symptomatique. Dans certains pays, dont l’Allemagne, le cabozantinib est également autorisé en cas de MTC évolutif, et est indiqué dans notre pays depuis 2017 pour le traitement du carcinome avancé des cellules rénales. En l’absence de preuve claire d’un bénéfice au stade précoce de la maladie, l’indication d’un traitement par ITK est actuellement considérée comme une maladie à charge tumorale élevée et progressive (critères RECIST, temps de doublement court des marqueurs tumoraux) ainsi que très symptomatique, lorsque les mesures thérapeutiques locales ont été épuisées. Aucune augmentation de la survie n’a été démontrée à ce jour avec un ITK dans le MTC métastatique [11].
Malgré les métastases à distance, de nombreux patients ont une bonne qualité de vie pendant des années, car la croissance de la tumeur est souvent très lente. Une stratégie de “surveillance active” est possible. Le traitement de la MTC métastatique est résolument axé sur les symptômes, y compris le traitement antidiarrhéique systématique au stade avancé de la tumeur avec du lopéramide et/ou de la tinctura opii, souvent négligé ou dosé trop prudemment au détriment de la qualité de vie.
Suivi du MEN2
En plus du suivi du MTC, un diagnostic concernant le phéochromocytome est effectué chaque année à partir de 11 ans pour les mutations RET à risque élevé et maximal et à partir de 16 ans pour les mutations RET à risque modéré, au moyen de dosages de métanéphrine et de catécholamines et, si nécessaire, d’une imagerie IRM. En ce qui concerne l’hyperparathyroïdie primaire, un contrôle annuel avec dosage du calcium sérique et de la parathormone est effectué de manière analogue en ce qui concerne l’âge dans les groupes de risque de mutation correspondants (tableau 1).
Messages Take-Home
- Le marqueur tumoral du carcinome médullaire de la thyroïde (CMT) est la calcitonine.
- Le dépistage de la calcitonine en cas de goitre nodulaire permet un diagnostic précoce de la MTC.
- Tous les patients atteints de MTC doivent faire l’objet d’une analyse génétique moléculaire du proto-oncogène RET au moment du diagnostic.
- En règle générale, le traitement de choix est la thyroïdectomie totale avec lymphadénectomie centrale.
- Dans le cadre du suivi, une distinction est faite entre les cas de guérison biochimique (Ctn non mesurable et bas), les cas de guérison biochimique incomplète (Ctn élevé sans détection de tumeur à l’imagerie) et les cas de détection de tumeur structurelle (métastases à l’imagerie).
- Dans de nombreux cas avec métastases, la “surveillance active” est possible ; en cas de progression et de symptômes, la règle est : local (chirurgie palliative, radiothérapie) avant systémique (inhibiteurs de tyrosine kinase).
Littérature :
- Wells SA Jr, et al : Révision des lignes directrices de l’Association américaine de thyroïde pour la prise en charge du carcinome médullaire de la thyroïde. Thyroïde 2015 ; 25(6) : 567-610.
- Ceolin L, et al : Carcinome médullaire de la thyroïde au-delà de la chirurgie : avancées, défis, et perspectives. Endocr Relat Cancer 2019 ; 26(9) : R499-R518.
- Raue F, Frank-Raue K : Dépistage de la calcitonine dans les limites supérieures de goitre nodulaire. Dtsch Arztebl Int 2018 ; 115(13) : 221.
- Costante G, et al : Détermination des niveaux de calcitonine dans la maladie à cellules C : intérêt clinique et pièges potentiels. Nat Clin Pract Endocrinol Metab 2009 ; 5(1) : 35-44.
- Kratzsch J, Petzold A, Raue F, et al : Calcitonine et procalcitonine basale et stimulée par divers tests chez des patients avec et sans cancer médullaire de la thyroïde. Clin Chem 2011 ; 57(3) : 467-474.
- Kahaly GJ, Algeciras-Schimnich A, Davis TE, et al : United States and European Multicenter Prospective Study for the Analytical Performance and Clinical Validation of a Novel Sensitive Fully Automated Immunoassay for Calcitonin. Clin Chem 2017 ; 63(9) : 1489-1496.
- Mian C, Perrino M, Colombo C, et al : Refining calcium test for the diagnosis of medullary thyroid cancer : cutoffs, procedures, and safety. J Clin Endocrinol Metab 2014 ; 99(5) : 1656-1664.
- Frank-Raue K, Schott M, Raue F. Au nom de la section de la thyroïde de la DGE. [Recommendation for Calcitonin Screening in Nodular Goiter]. Dtsch Med Wochenschr 2018 ; 143(15) : 1065-1069.
- Machens A, Dralle H. Biomarker-based risk stratification for previously untreated medullary thyroid cancer. J Clin Endocrinol Metab 2010 ; 95(6) : 2655-2663.
- Mucha L, Leidig-Bruckner G, Frank-Raue K, et al : Phaeochromocytoma in multiple endocrine neoplasia type 2 : RET codon-specific penetrance and changes in management during the last four decades. Clin Endocrinol (Oxf) 2017 ; 87(4) : 320-326.
- Cabanillas ME, Ryder M, Jimenez C : Thérapie ciblée pour les cancers avancés de la thyroïde : inhibiteurs de kinase et au-delà. Endocr Rev 2019 ; 40(6) : 1573-1604.
- Machens A, Gimm O, Ukkat J, et al : Improved prediction of calcitonin normalization in medullary thyroid carcinoma by quantitative lymph node analysis. Cancer 2000 ; 88(8) : 1909-1915.
- Roman S, Lin R, Sosa JA : Prognosis of medullary thyroid carcinoma : demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 2006 ; 107(9) : 2134-2142.
- Randle RW, Balentine CJ, Leverson GE et al : Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 2017 ; 161(1) : 137-146.
- Opsahl EM, Akslen LA, Schlichting E, et al. : Trends in Diagnostics, Surgical Treatment, and Prognostic Factors for Outcomes in Medullary Thyroid Carcinoma in Norway : A Nationwide Population-Based Study. Eur Thyroid J 2019 ; 8(1) : 31-40.
- Lindsey SC, Ganly I, Palmer F, et al : Response to initial therapy predicts clinical outcomes in medullary thyroid cancer. Thyroïde 2015 ; 25(2) : 242-249.
- Yang JH, Lindsey SC, Camacho CP, et al : L’intégration d’une mesure postopératoire de la calcitonine dans un système de staging anatomique améliore la stratification du risque initial dans le cancer médullaire de la thyroïde. Clin Endocrinol (Oxf) 2015 ; 83(6) : 938-942.
- Raue F, Frank-Raue K : Suivi à long terme du carcinome médullaire de la thyroïde. Résultats récents Cancer Res 2015 ; 204 : 207-225.
- Leidig-Bruckner G, Bruckner T, Raue F, et al : Long-Term Follow-Up and Treatment of Postoperative Permanent Hypoparathyroidism in Patients with Medullary Thyroid Carcinoma : Differences in Complete and Partial Disease. Horm Metab Res 2016.
- www.thyroid.org/professionals/calculators/thyroid-cancer-carcinoma
- Laure Giraudet A, Al Ghulzan A, Auperin A, et al : Progression du carcinome médullaire de la thyroïde : évaluation avec la calcitonine et les temps de doublement de l’antigène carcinoembryonnaire. Eur J Endocrinol 2008 ; 158(2) : 239-246.
- Hadoux J, Pacini F, Tuttle RM, et al : Prise en charge du cancer médullaire avancé de la thyroïde. Lancet Diabetes Endocrinol 2016 ; 4(1) : 64-71.
- Vogel T, Wendler J, Frank-Raue K, et al : Métastases osseuses dans le carcinome médullaire de la thyroïde : morbidité élevée et mauvais pronostic associés à une morphologie ostéolytique. J Clin Endocrinol Metab 2020 ; 105(6).
InFo ONKOLOGIE & HÉMATOLOGIE 2020 ; 8(3) : 6-10