L’apparition d’une insuffisance cardiaque à fraction d’éjection préservée (HFpEF) est typique de l’amylose cardiaque. De nouvelles méthodes de diagnostic et de nouvelles possibilités thérapeutiques jettent désormais un éclairage différent sur cette maladie rare et pratiquement incurable jusqu’à présent.
Avec l’introduction de nouvelles méthodes de diagnostic et de nouvelles possibilités thérapeutiques pour une maladie rare pratiquement incurable jusqu’à présent, l’intérêt pour l’amylose croît rapidement. L’atteinte cardiaque (amylose cardiaque) entraîne généralement une insuffisance cardiaque grave avec toutes ses conséquences cliniques. L’objectif de cet article est de donner un bref aperçu du diagnostic et du traitement modernes de l’amylose cardiaque.
Origine
L’amylose systémique se caractérise par des dépôts de protéines anormalement repliées et agrégées dans les tissus. En fonction de l’atteinte des organes, les troubles fonctionnels et les maladies correspondantes se présentent. Si les dépôts sont dans le cœur (dans l’interstitium entre les cellules du myocarde), cela entraîne une maladie cardiaque avec un tableau clinique d’insuffisance cardiaque. Deux types d’amyloïdes différents se déposent généralement dans le cœur, en fonction de la protéine anorexigène défectueuse.
Amylose AL : elle se développe généralement sur le terrain d’une dyscrasie plasmocytaire. Les cellules plassma clonales (par exemple dans le cas d’un myélome multiple) produisent un excès de chaînes légères libres. Dans le cas des chaînes légères amyloïdogènes, elles se déposent sous forme de fibrilles amyloïdes dans différents organes. Dans l’amylose AL, l’atteinte cardiaque isolée est rare. Le plus souvent, les reins, le foie et le système nerveux périphérique sont également touchés. Les chaînes légères lambda sont beaucoup plus souvent détectées comme composant amyloïde que les chaînes kappa.
Amylose ATTR : La transthyrétine (TTR) est une protéine de transport produite à 98% dans le foie et responsable du transport de la thyroxine et du rétinol. Le TTR est un tétramère qui peut se dissocier en quatre monomères. Normalement, ces monomères sont solubles dans le sang. Cependant, en cas de dysfonctionnement, les monomères s’agrègent pour former des fibrilles amyloïdes pathologiques. Il existe deux causes d’amylose TTR : la forme mutée (héréditaire, familiale) est due à une mutation du gène TTR. La prévalence des mutations varie selon les régions géographiques et les groupes ethniques. La forme “wild-type” (acquise, sénile) se manifeste généralement à un âge avancé et affecte principalement le cœur.
Manifestation clinique
Atteinte cardiaque : L’apparition d’une insuffisance cardiaque à fraction d’éjection préservée (HFpEF) est typique de l’amylose cardiaque. Les dépôts amyloïdes rendent les parois des ventricules et des oreillettes plus épaisses (pas d’hypertrophie à proprement parler, car les cellules du muscle cardiaque ne sont pas affectées en soi). Cela entraîne une “rigidification” du cœur et une limitation de sa relaxation (dysfonction diastolique). Il en résulte une augmentation des pressions de remplissage diastolique dans le ventricule, une dilatation des oreillettes et une augmentation de la pression pulmonaire. Cliniquement, cela se traduit finalement par une insuffisance cardiaque gauche et droite avec dyspnée, intolérance à l’effort et œdèmes. Il est également typique que le ventricule rigide ne puisse plus adapter correctement le volume des minutes cardiaques. En raison du volume de battement fixé par la rigidité, le débit cardiaque ne peut pratiquement plus être contrôlé que par la fréquence cardiaque. Des syncopes et une dérégulation orthostatique sévère peuvent donc survenir, en particulier à l’effort, surtout chez les patients sous bêtabloquants.
L’augmentation de la taille de l’oreillette gauche est un “bon” substrat pour le développement de la fibrillation auriculaire. Des pressions élevées, associées à une oreillette peu mobile en raison des dépôts amyloïdes, peuvent entraîner un risque accru de thrombus intra-atrial et expliquent le risque nettement accru d’infarctus cérébral chez les patients atteints d’amylose.
Si les dépôts amyloïdes affectent le système de conduction, des blocages se produisent, en particulier des blocages AV. En outre, le risque de mort subite par troubles du rythme ventriculaire (notamment fibrillation ventriculaire et activité électrique sans pouls – PEA) est accru.
Une autre manifestation typique de l’amylose cardiaque est la douleur thoracique, sans qu’une maladie coronarienne proprement dite puisse être trouvée. D’une part, de petites microthromboses dans la microcirculation peuvent déclencher une angine de poitrine (spasmes), d’autre part, la perturbation de la fonction endothéliale révèle un dysfonctionnement microvasculaire.
Atteinte d’autres organes : le problème fréquent d’orthostatisme chez les patients atteints d’amylose est généralement dû à l’atteinte du système nerveux périphérique et à la diminution de la vasoconstriction des vaisseaux périphériques qui en résulte.
Un dysfonctionnement de l’innervation intestinale peut entraîner des troubles du transit, des ballonnements, des douleurs abdominales et une irrégularité des selles. L’atteinte rénale se traduit généralement par une protéinurie et une diminution de la fonction rénale.
L’accumulation d’amyloïde dans le tractus gastro-intestinal entraîne parfois une malabsorption, une perte de poids et une augmentation de la rigidité du foie. Dans l’amylose AL, on peut parfois (10%) trouver les signes pathognomoniques d’une hémorragie périorbitaire et d’une macroglossie. L’amylose TTR est souvent précédée d’un syndrome du canal carpien.
Diagnostic
En cas de suspicion clinique d’amylose cardiaque, de nombreuses méthodes de diagnostic sont utilisées. L’ECG révèle souvent un faible voltage périphérique (affaiblissement de la conduction électrique vers les électrodes en raison du dépôt d’amyloïde), un signe distinctif par rapport à la cardiomyopathie hypertrophique, où l’on observe généralement un voltage accru. Il est également possible de voir des images de bloc, de fibrillation auriculaire et d’autres troubles du rythme. Tous ces changements ne sont toutefois pas spécifiques à la maladie. En laboratoire, outre les paramètres courants, les biomarqueurs cardiovasculaires NT-proBNP et troponine doivent notamment être mesurés. Une analyse d’urine, avec notamment la question de l’albumine et de l’urine, ne doit pas non plus être omise. Une électrophorèse des protéines et une immunofixation dans le sérum et l’urine sont également indispensables pour distinguer les formes AL et ATTR.
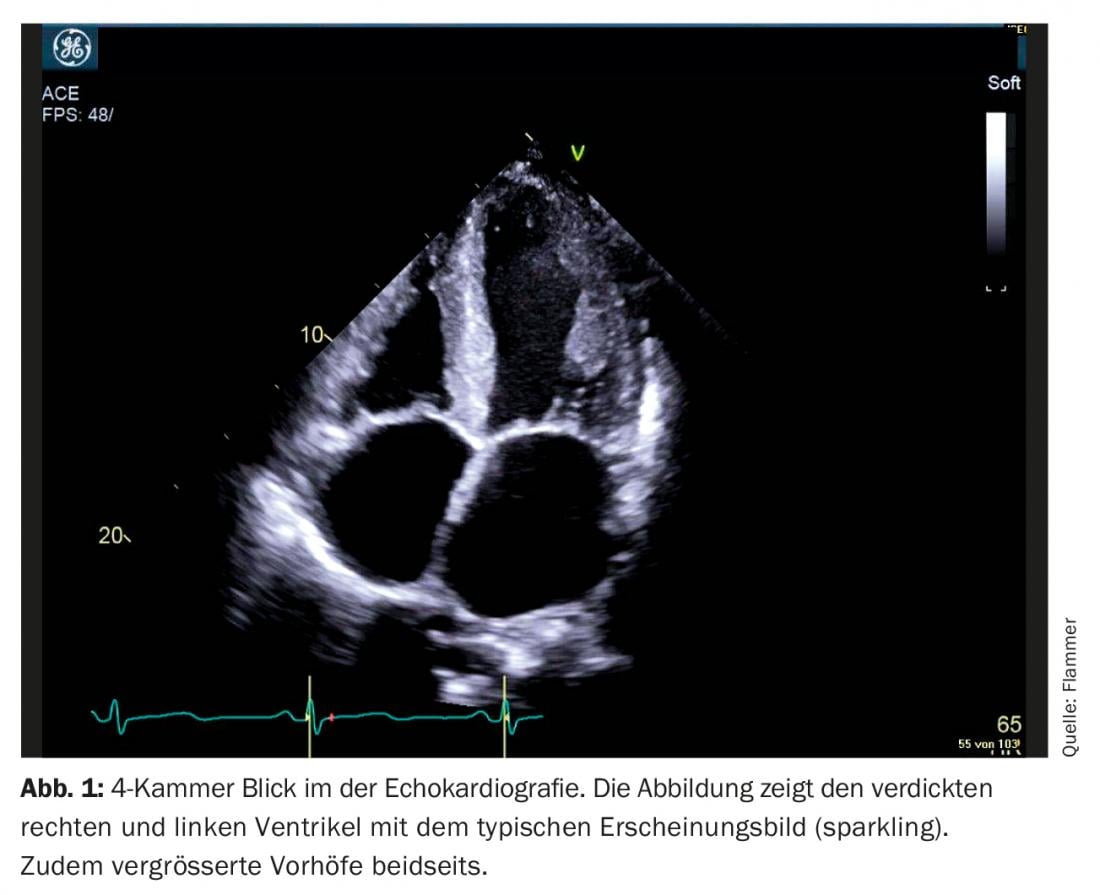
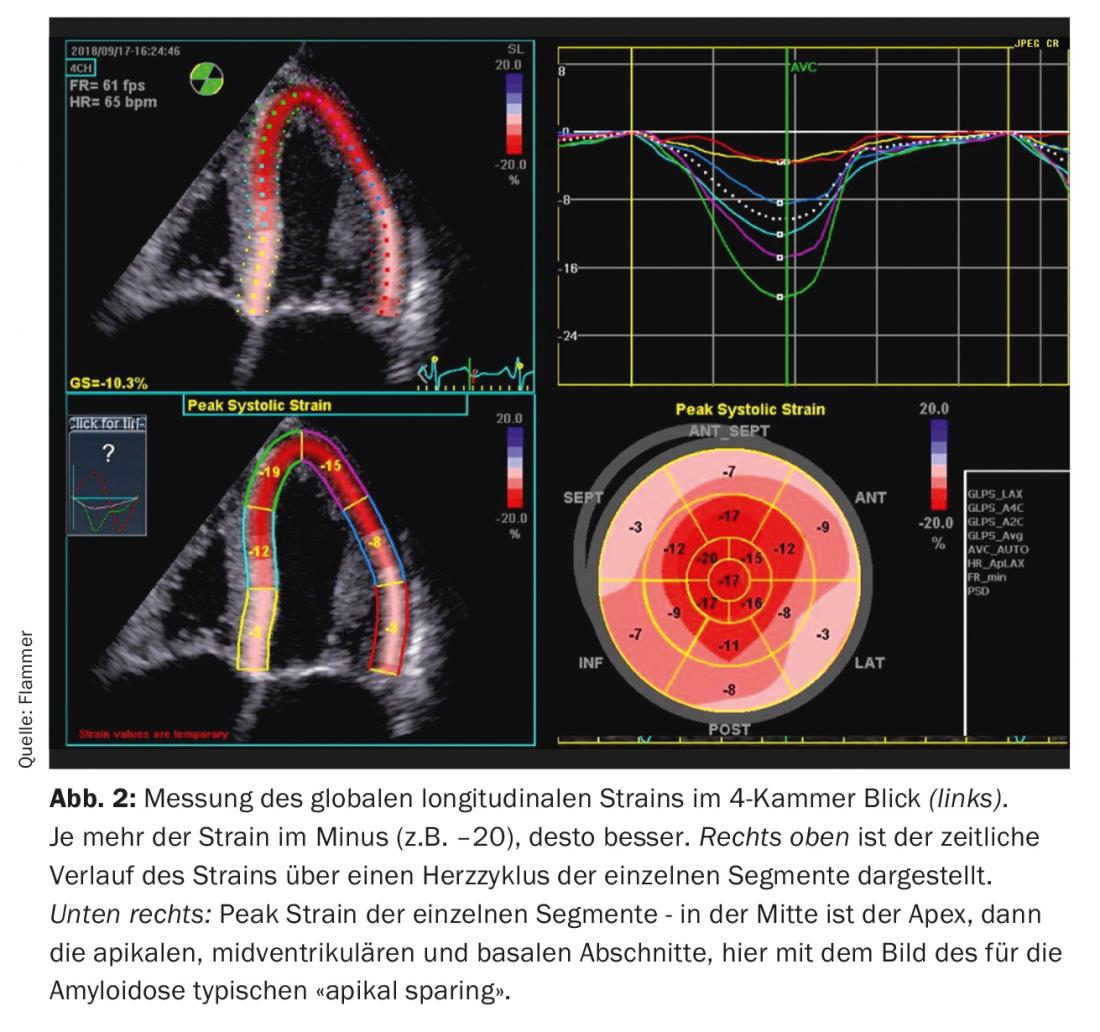
Echocardiographie : le diagnostic d’amylose cardiaque est impensable sans imagerie cardiaque. Les patients présentant des antécédents et des signes d’insuffisance cardiaque subissent généralement une échocardiographie. L’amylose doit être évoquée en présence de signes d’amylose correspondants à l’écho et d’une insuffisance cardiaque manifeste. Quels sont les signes typiques de l’échocardiographie ? On observe un épaississement du myocarde biventriculaire, le septum intra-auriculaire et les parois auriculaires sont également généralement épaissis. Le myocarde présente également un aspect particulier “scintillant”, caractéristique de la maladie (il convient toutefois de noter que la fonction “second harmonic imaging” doit être désactivée sur l’échographe). L’amyloïde se dépose également sur les valves. Ainsi, elles apparaissent légèrement épaissies, mais le fonctionnement des valves est généralement normal. Un épanchement péricardique discret est également souvent observé (Fig. 1). Sur le plan fonctionnel, on observe, surtout à un stade avancé de la maladie, un dysfonctionnement diastolique sévère, généralement associé à un schéma de remplissage restrictif. En raison de l’augmentation des pressions diastoliques qui en résulte, les deux oreillettes augmentent de volume (dilatation biatriale) au cours de la maladie, ce qui est caractéristique d’un dysfonctionnement diastolique sévère. Bien que la fraction d’éjection soit généralement normale, on observe également une dysfonction systolique qui se traduit par une diminution du “strain” (le strain décrit la déformation mesurée du myocarde). Ainsi, le “strain” longitudinal global est notamment limité. L’amylose se caractérise par ce que l’on appelle l'”apical sparing”, c’est-à-dire que le strain est normal (évidé) à l’apex, mais qu’il se détériore au fur et à mesure que l’on se dirige vers la base du cœur (Fig. 2).
Imagerie par résonance magnétique (IRM) : de nos jours, l’amylose est souvent diagnostiquée par cet examen (parfois comme découverte fortuite dans le cadre d’autres questions). Les modifications observées à l’IRM sont en principe similaires à celles observées à l’écho, mais d’autres modifications typiques peuvent être observées, en particulier lorsque le produit de contraste gadolinium est également utilisé. En raison d’un “wash-out” lent du gadolinium, celui-ci reste plus longtemps dans l’interstitium s’il y a de l’amyloïde. L’IRM révèle alors un “rehaussement tardif du gadolinium” diffus et en forme de tache, typique de l’amylose.
Algorithme de diagnostic en cas de suspicion d’amylose cardiaque
Chez les patients présentant une clinique correspondante et une suspicion d’amylose cardiaque à l’imagerie, il convient d’exclure en premier lieu une dyscrasie plasmocytaire. Pour cela, il faut rechercher une électrophorèse des protéines et une immunofixation dans l’urine et le sérum, comme décrit ci-dessus. Si elle est présente, on recherchera en premier lieu une amylose AL, et en l’absence de signes, une amylose ATTR (figure 3).
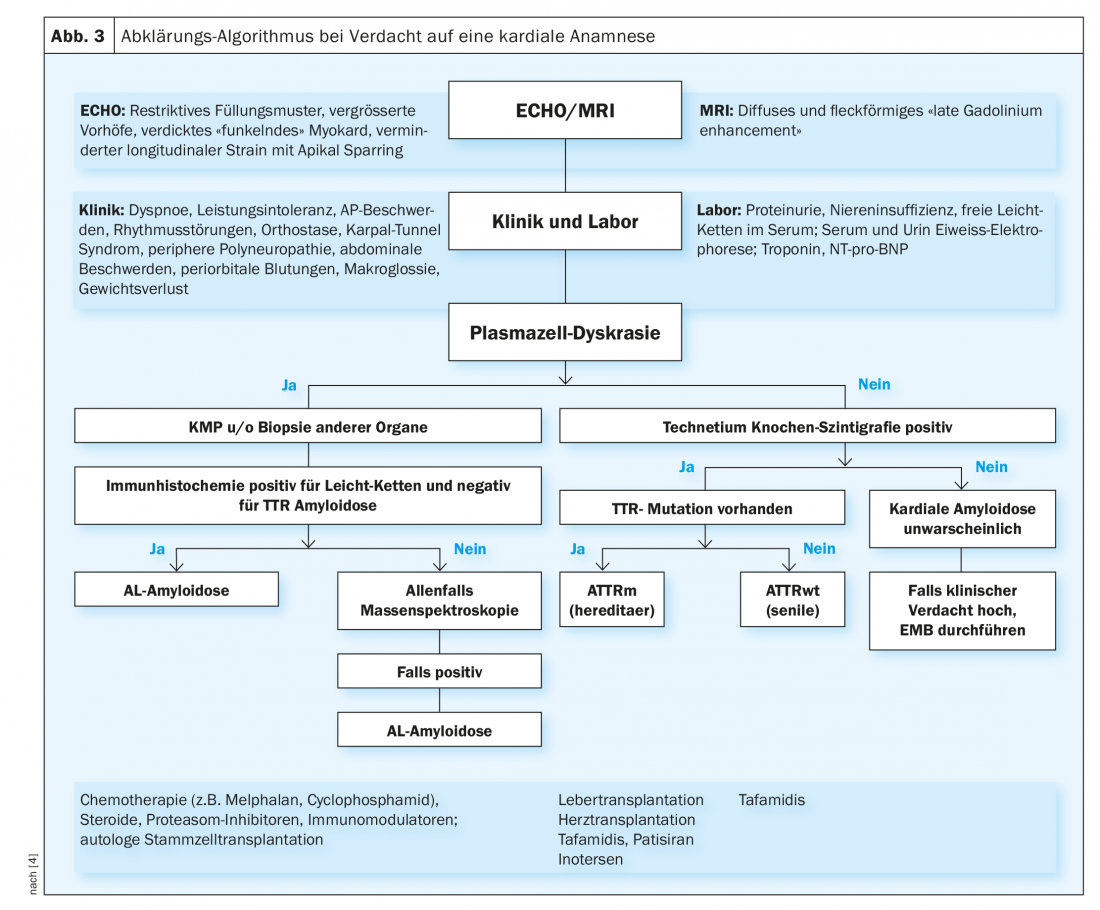
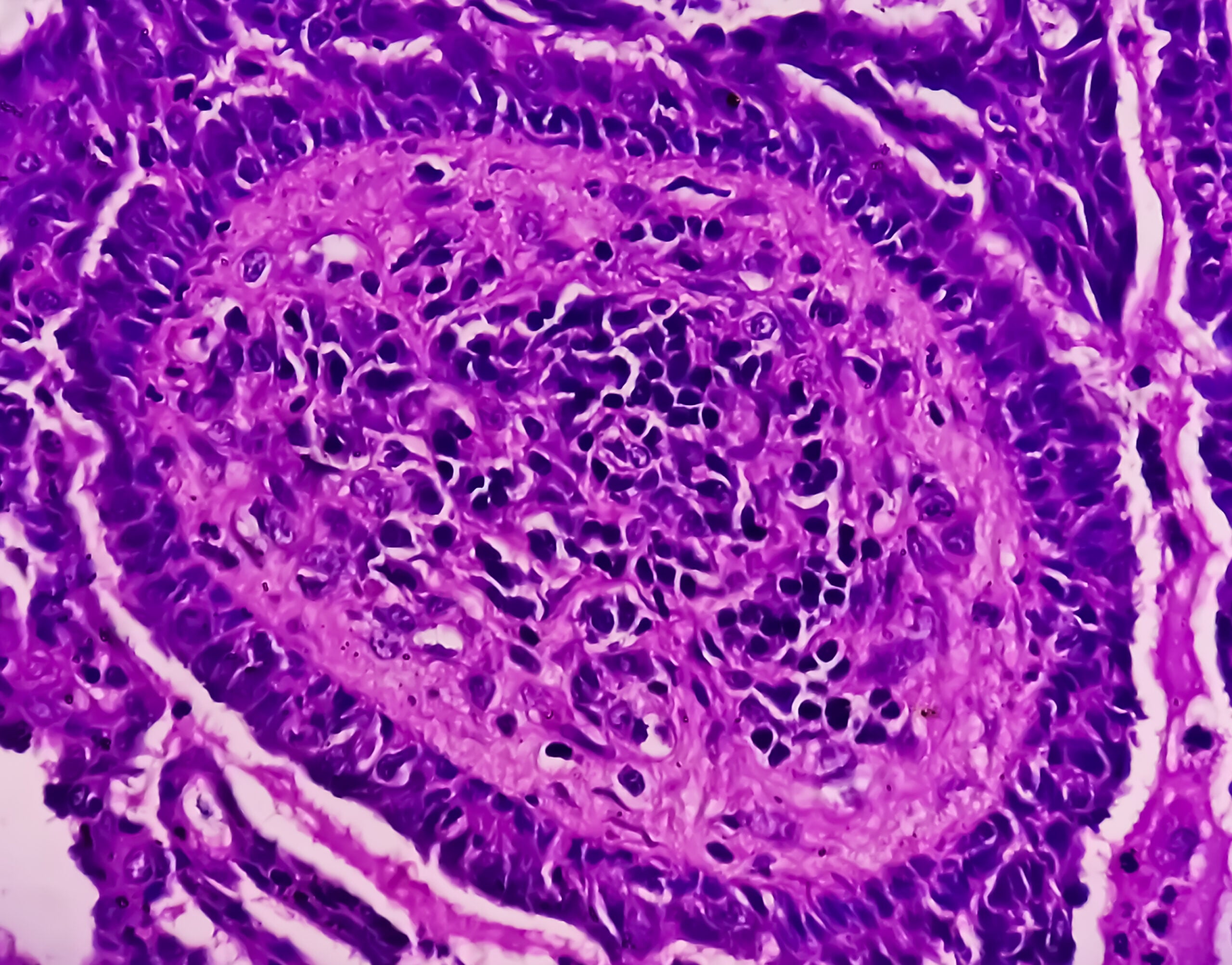
En cas de suspicion d’amylose AL (présence d’une dyscrasie plasmocytaire), il faut essayer de détecter l’amyloïde AL directement par biotopage. Le plus souvent, une ponction de la moelle osseuse est effectuée pour diagnostiquer la maladie hématologique sous-jacente (généralement un myélome multiple). L’amylose étant une maladie systémique, le diagnostic est confirmé dès qu’une biopsie (de n’importe quel tissu) révèle la présence d’AL-amyloïde. L’aspiration de tissu adipeux abdominal est la moins invasive et la moins traumatisante. De même, l’amyloïde peut parfois être recherchée dans des prélèvements de tissus effectués précédemment dans le cadre d’une coloscopie ou d’une gastroscopie. Des biopsies des glandes salivaires, des lèvres ou du rectum peuvent également être réalisées. Parfois, une biopsie rénale ou endomyocardique est également nécessaire pour établir un diagnostic correct. Dans le cas de l’amylose cardiaque, la biopsie endomyocardique est très sensible. D’une part, les biopsies sont analysées pour détecter la présence d’amyloïde et, d’autre part, l’immunohistochimie permet d’essayer de classer le type d’amylose. Dans les cas difficiles, une spectroscopie de masse (Goldstandart) est nécessaire. Cependant, rares sont les pathologies qui réalisent un tel examen.
En cas de suspicion d’amylose TTR (absence de signes de dyscrasie plasmocytaire), une scintigraphie au technétium (os) est aujourd’hui réalisée. Dans ces conditions, la sensibilité et la spécificité de cette méthodologie sont très bonnes pour l’amylose ATTR cardiaque. En cas de résultat positif, il est donc possible de renoncer à une biopsie et de poser le diagnostic. Si la scintigraphie n’est pas claire et que la suspicion d’amylose cardiaque persiste, une biopsie endomylocardique doit être réalisée à ce stade.
Une fois l’amylose ATTR confirmée, une analyse génétique est nécessaire pour distinguer la forme “sauvage” de la forme héréditaire.
Traitement de l’amylose cardiaque
L’insuffisance cardiaque est généralement au centre de la symptomatologie de l’amylose cardiaque. Il faut les aborder en premier lieu sur le plan thérapeutique. Les diurétiques sont une aide symptomatique. En particulier parce qu’en raison de la physiologie cardiaque restrictive, les pressions de remplissage cardiaques sont élevées et entraînent une augmentation de la pression pulmonaire veineuse passive. Sous diurétiques, les patients sont nettement moins symptomatiques. Au fur et à mesure de l’évolution de la maladie, des doses de plus en plus élevées sont généralement nécessaires – il faut donc également veiller à ce que la substitution en potassium soit suffisante. La titration de la dose est souvent un défi (encadré).

Conformément à la physiologie HFpEF, il n’y a pas d’indication pour les inhibiteurs de l’ECA, les bêtabloquants et les antagonistes de l’aldostérone. Au contraire, les inhibiteurs de l’ECA en particulier peuvent renforcer une orthostatisme souvent déjà prononcé. Les bêtabloquants ont pour effet d’empêcher l’augmentation du débit cardiaque à l’effort en raison de la fixité du volume de battements. Ainsi, les bêtabloquants sont réservés aux patients en fibrillation auriculaire tachycardique.
La fibrillation auriculaire et les infarctus cérébraux qui en résultent sont fréquents. Il est d’une part essentiel de ne pas manquer l’HVF (examens Holter tous les 6 mois), et d’autre part d’instaurer une anticoagulation orale en cas de présence, et ce indépendamment du score vasculaire CHADS. Il n’est pas totalement établi si un traitement OAK doit être initié en l’absence de FVC – cela est probablement utile en cas de schéma de remplissage restrictif. En cas de tachycardie VHF, un contrôle de la fréquence doit être effectué avec un bêtabloquant, la digoxine ne doit pas être administrée en raison d’une toxicité accrue. L’amiodarone est indiquée chez certains patients pour le contrôle du rythme et occasionnellement pour le contrôle de la fréquence.
En raison des troubles du système de conduction cardiaque, l’implantation d’un stimulateur cardiaque est nécessaire, en particulier chez les patients atteints d’amylose TTR, une indication qui peut être posée de manière généreuse. La question de l’implantation d’un défibrillateur cardioverteur implantable (DAI) constitue un défi particulier. La cause de décès cardiaque la plus fréquente chez les patients atteints d’amylose est la mort subite d’origine cardiaque due à des troubles du rythme ventriculaire. On observe non seulement une fibrillation ventriculaire, mais aussi un nombre pertinent de PEA. Ces derniers ne sont pas détectés de manière fiable par un DAI. Il y a également un taux plus élevé de chocs appliqués par erreur (ce qui est très traumatisant pour les patients) et de chocs non réussis. Jusqu’à présent, il n’existe que des études rétrospectives sur ce sujet, qui ne montrent pas de bénéfice clair pour un DAI. L’indication doit donc être posée avec réserve et discutée en détail au sein d’une équipe spécialisée dans l’amylose.
Thérapie causale
Dans le cas de l’amylose AL, il faut s’attaquer en premier lieu à la maladie hématologique. Cependant, cela représente souvent un défi, en particulier en cas d’atteinte cardiaque sévère et d’insuffisance cardiaque grave concomitante. Un traitement à haute dose par autogreffe de cellules souches, souvent envisagé, ne peut pas être réalisé dans ce cas. Les autres schémas thérapeutiques avec des agents chimiothérapeutiques (p. ex. melphalan, cyclophosphamide), des stéroïdes et de nouveaux inhibiteurs du protéasome, des immunomodulateurs ou des anticorps anti-CD38 peuvent également être occasionnellement stressants pour le cœur. Toutefois, dans certains cas, il existe des approches curatives. Mais une étude détaillée dépasserait le cadre de cet article.
Pour l’amylose TTR héréditaire, la transplantation hépatique (et, à un stade avancé, la transplantation cardiaque) reste le traitement de choix.
Cependant, depuis l’été 2018, deux médicaments sont disponibles aux États-Unis et en Europe (autorisation de la FDA et de l’EMA). Ces médicaments utilisent la technologie d’interférence ARN pour ‘rendre muet’ le gène défectueux et ainsi éviter la production de TTR amyloïde. Ces deux médicaments ne sont pas encore autorisés en Suisse. Une étude à grande échelle chez des patients atteints d’amylose héréditaire a montré de manière impressionnante comment la polyneuropathie (qui prédomine souvent dans l’amylose héréditaire) peut être stoppée. Des analyses de sous-groupes pour le médicament patisiran montrent que les résultats pourraient être aussi bons au niveau cardiaque, mais il faut attendre d’autres études. Jusqu’à récemment, il n’existait aucun traitement causal pour l’amylose TTR de type sauvage. Heureusement, une étude également publiée à l’été 2018 a montré que le tafamidis, un stabilisateur de transthyrétine également autorisé en Europe et aux États-Unis pour le traitement de la névrite amyloïde héréditaire, donnait de bons résultats dans l’amylose cardiaque, avec une amélioration de la qualité de vie, une réduction de la mortalité et une diminution des hospitalisations d’origine cardiaque. Le médicament n’est actuellement pas encore disponible en Suisse, mais l’autorisation de mise sur le marché est attendue prochainement en raison des bons résultats obtenus.
Messages Take-Home
- L’amylose AL est une maladie rare qui est généralement causée par une maladie hématologique sous-jacente “produisant des chaînes légères” et qui affecte souvent d’autres organes en plus du cœur. Le traitement de la maladie hématologique est au cœur de la thérapie.
- L’amylose TTR peut être causée par une mutation du gène TTR ou être acquise au cours de la vie (“type sauvage”). La forme familiale, plus rare, s’exprime de manière prédominante par une polyneuropathie ou une cardiopathie (ou une combinaison des deux), selon l’atteinte génétique, tandis que la forme “sauvage” est généralement limitée au cœur. Outre le traitement symptomatique, il existe de nouvelles approches thérapeutiques prometteuses utilisant des stabilisateurs TTR et des molécules d’interférence ARN.
- Le diagnostic d’amylose cardiaque nécessite une clinique appropriée (insuffisance cardiaque), une échographie ou une IRM typique de l’amylose et soit une détection de l’amyloïde dans une biopsie, soit une scintigraphie Tc positive.
- La clinique typique de l’amylose cardiaque comprend notamment des symptômes d’insuffisance cardiaque, d’orthostatisme, de syncopes, de troubles du rythme (principalement HVF et tachycardie/fibrillation ventriculaire), de blocages AV et d’angor microvasculaire.
Littérature :
- Brouwers S, et al : Cardiac Amyloidosis Cardiovasc Med. 2018 ; 21(11) : 282-289.
- Rauch PJ, et al : Amyloïdoses systémiques Suisse Med Forum 2014 14 ; 943-948.
- Laptseva N, et al : Cardiac amyloidosis : still challenging Eur Heart J 2017, 38(22) : 122.
- Falk RH, et al. : AL (Light-Chain) Cardiac Amyloidosis : A Review of Diagnosis and Therapy.
- Gillmore JD, et al : Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis Circulation 2016 : j133(24) : 2404-2412.
- Maurer MS, et al : Tafamidis Tratment for Patients with Transthyretin Amyloid Cardiomyopath N Engl J Med 2018 ; 379(11) : 1007-1016.
- Adams D, et al : Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis N Engl J Med 2018 ; 379(1) : 11-21.
CARDIOVASC 2019 ; 18(2) : 6-10