L’atteinte pulmonaire dans le cadre d’une maladie rhumatismale inflammatoire est une manifestation relativement fréquente qui nécessite des connaissances appropriées tant chez les pneumologues que chez les rhumatologues et, idéalement, une prise en charge interdisciplinaire de ces cas. On y trouve diverses pathologies pulmonaires parenchymateuses, dont les altérations granulomateuses constituent un sous-groupe.
L’atteinte pulmonaire dans le cadre d’une maladie rhumatismale inflammatoire est une manifestation relativement fréquente qui nécessite des connaissances appropriées tant chez les pneumologues que chez les rhumatologues et, idéalement, une prise en charge interdisciplinaire de ces cas. On y trouve diverses pathologies pulmonaires parenchymateuses, dont les altérations granulomateuses constituent un sous-groupe. Le terme granulome désigne une accumulation circonscrite en forme de nodule de cellules inflammatoires dans les tissus, classiquement des macrophages, mais aussi des granulocytes ou des lymphocytes. En cas de preuve histologique d’une pneumopathie granulomateuse, plusieurs diagnostics différentiels rhumatologiques doivent être traités. L’objectif de cet article est de donner un aperçu des maladies pulmonaires granulomateuses dans le domaine de la rhumatologie, à l’exception de la sarcoïdose. L’accent est mis sur le diagnostic et surtout sur la thérapie.
Vascularites associées aux ANCA
Les vascularites associées aux ANCA sont les premières à entrer en ligne de compte. Ce groupe de vascularite des petits vaisseaux comprend trois entités : La granulomatose avec polyangéite (GPA), la polyangéite microscopique (MPA) et la granulomatose éosinophile avec polyangéite (EGPA), la MPA n’étant pas susceptible de former des granulomes à l’histologie.
Granulomatose avec polyangéite (GPA), tableau clinique
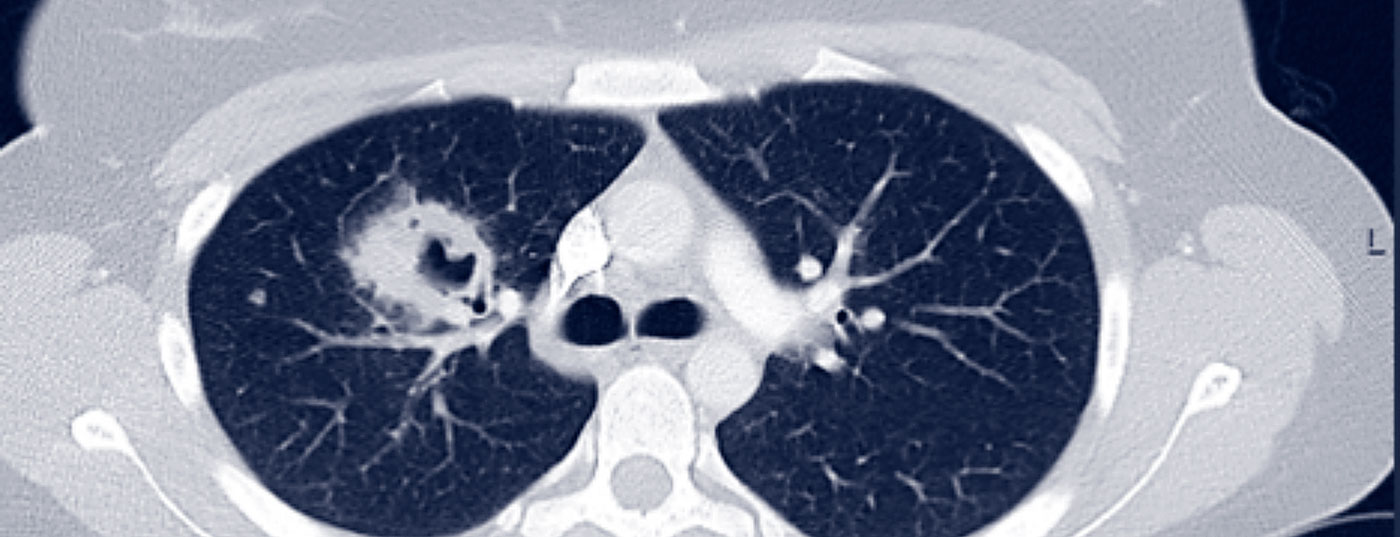
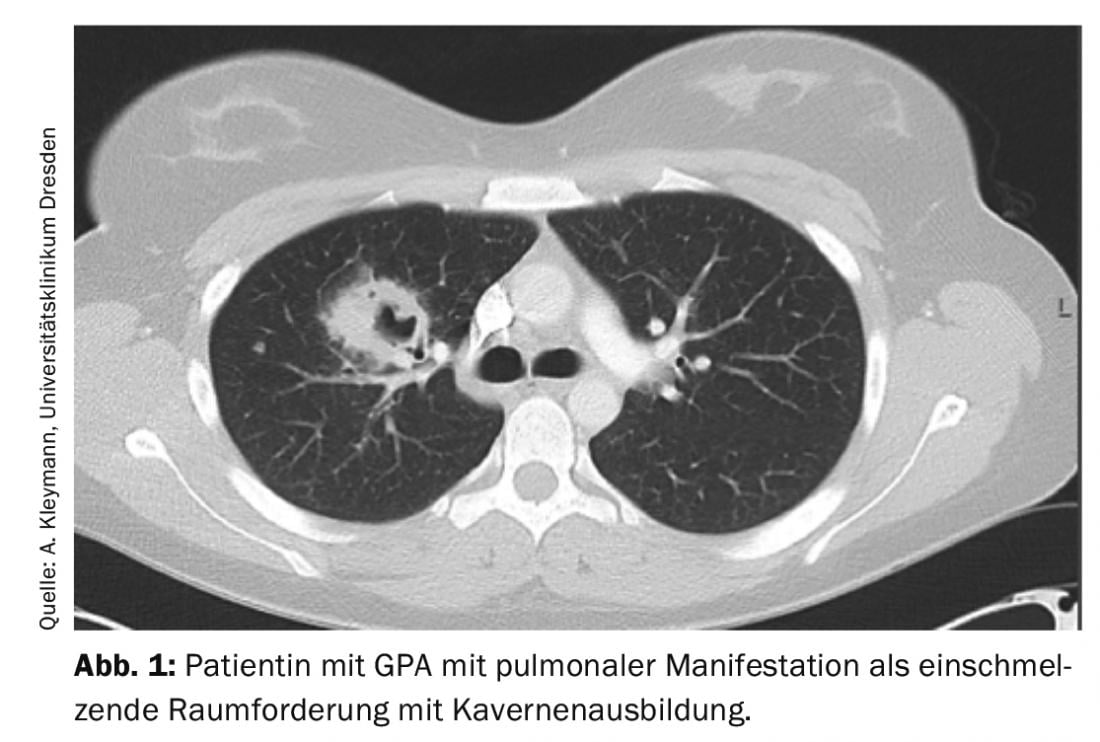
L’AMP peut être initialement oligosymptomatique (stade localisé de la maladie). La première manifestation typique est l’atteinte du nasopharynx dans le sens d’une sinusite ou d’une rhinite chronique. En cas d’atteinte pulmonaire, la GPA est déjà considérée comme une maladie généralisée, de sorte que l’on peut typiquement s’attendre à une symptomatologie clinique (symptômes B, myalgies, arthralgies, éruptions cutanées, arthrite) et paraclinique (augmentation de la CRP et de la BSG, anémie, leucocytose, thrombocytose) concomitante. L’atteinte pulmonaire peut prendre la forme de nodules pulmonaires, d’infiltrats, de fibrose pulmonaire, de cavernes ou encore d’hémorragies alvéolaires (figure 1).
Diagnostic ANCA
Un élément important du diagnostic est la détermination des ANCA (anticorps cytoplasmiques antineutrophiles) avec des auto-anticorps dirigés contre la protéinase-3 et la myéloperoxydase. Les ANCA ont été décrits pour la première fois en 1982 dans la glomérulonéphrite et ont été initialement considérés comme associés à un virus. Ce n’est qu’en 1985 qu’il a été décrit chez les patients atteints d’AMP. La détermination par immunofluorescence indirecte est initialement recommandée comme gold standard. Les résultats doivent être classés en fonction du modèle de fluorescence et du titre. Le pANCA est un motif de fluorescence périnucléaire, tandis que le cANCA est un motif cytoplasmique (Fig. 2). Parfois, le motif de fluorescence est indiqué comme xANCA, mais il faut alors supposer qu’il s’agit d’ANCA non spécifiques, par exemple dans le cadre d’une colite ulcéreuse.
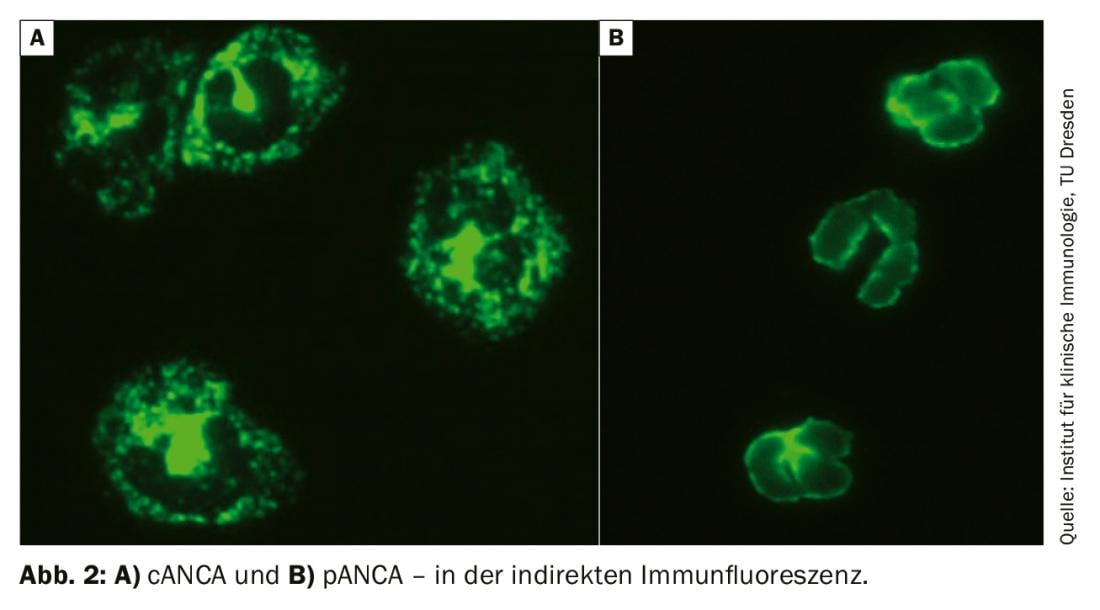
La spécificité des résultats ANCA positifs n’est augmentée que par la détection d’auto-anticorps dirigés contre les antigènes cibles correspondants, la myéloperoxydase pour le pANCA et la protéinase-3 pour le cANCA. Leur détermination se fait généralement par ELISA (Enzyme-linked Immunosorbent Assay). En cas de suspicion exprimée d’une vascularite associée aux ANCA, un dépistage structuré doit être mis en place concernant d’éventuelles autres manifestations organiques. Il faut notamment penser à une implication rénale sous la forme d’une glomérulonéphrite pauci-immune. C’est pourquoi, outre les paramètres de rétention (créatinine, stase urinaire, DFG), un sédiment urinaire doit être demandé sans exception. En cas d’érythrocyturie, de protéinurie ou de mise en évidence de cylindres hyalins, il faut demander une microscopie des urines en s’interrogeant sur la présence d’érythrocytes dysmorphiques dans le sens d’un sédiment néphritique actif et une quantification des protéines. En cas de suspicion confirmée de manifestation rénale, une biopsie rénale est nécessaire. Cela permet souvent non seulement de confirmer le diagnostic, mais aussi de déterminer le pronostic concernant la fonction rénale. Chez les patients obèses ou présentant un risque accru de saignement, la biopsie rénale transjugulaire constitue une alternative valable à la biopsie transcutanée.
Thérapie
Après le diagnostic, en cas de manifestation pulmonaire de la vascularite, un traitement d’induction par cyclophosphamide ou rituximab est généralement indiqué en accompagnement des corticostéroïdes, les corticostéroïdes devant être réduits assez rapidement, de sorte que la dose soit de 7,5 à 10 mg/j d’équivalent prednisolone après 3 mois. L’exception à cette règle est une manifestation isolée de petits nodules pulmonaires non désintégrés sans limitation fonctionnelle, où un traitement de base par méthotrexate, à condition qu’il n’y ait pas de limitation de la fonction rénale, ou par azathioprine est également possible sans traitement d’induction préalable.
Sur la base des données de l’étude CYCLOPS, l’induction par cyclophosphamide en bolus i.v. doit être privilégiée par rapport à la voie orale en raison des faibles taux d’effets indésirables [1]. La posologie standard est de 15 mg/kg de poids corporel, les 3 premières administrations se font toutes les 2 semaines, puis toutes les 3 semaines. La dose doit être ajustée en fonction de l’âge et de la fonction rénale (réduction à 12,5 mg/kgKG pour les personnes âgées de 60 à 70 ans si la créatinine atteint 300 µmol/l, à 10 mg/kgKG si la créatinine >300 µmol/l et chez les >70 ans si la créatinine est inférieure ou égale à 300 µmol/l, et à 7,5 mg/kgKG chez les >70 ans avec une créatinine >300 µmol/l). La plupart du temps, 6 bolus sont suffisants. Sous traitement par cyclophosphamide, outre la toxicité gonadique et urinaire (c’est pourquoi l’administration de 2-mercaptoéthanesulfonate de sodium (MESNA) est également recommandée), il convient de souligner le risque de leucopénie importante. Le nadir apparaît 10 à 12 jours après l’administration, de sorte qu’un contrôle approprié de l’hémogramme doit être effectué autour de cette période et, si nécessaire, une nouvelle adaptation de la dose pour l’administration suivante (si les leucocytes <4 GPT/l, réduction de la dose de cyclophosphamide de 25% et l’administration suivante seulement lorsque le nombre de leucocytes >4 GPT/l). Une alternative au cyclophosphamide est l’induction avec le rituximab, un anticorps anti-CD20. L’efficacité du rituximab a été démontrée dans 2 études (RAVE et RITUXVAS). Dans ce contexte, , le schéma consistant à administrer 4 fois par semaine 375 mg/m2 de surface corporelle ainsi que 2 fois 1 g de dose absolue à 14 jours d’intervalle est une pratique courante. Avant l’instauration du traitement par rituximab, il convient de procéder à un dépistage de l’hépatite (risque de réactivation de l’hépatite B chronique) et, idéalement, de mettre à jour le statut vaccinal, car il est probable que la réponse vaccinale soit réduite/insuffisante sous traitement.
Il ne faut pas sous-estimer le risque accru d’infection par des agents pathogènes atypiques avec les deux médicaments, de sorte qu’une prophylaxie de la pneumocystis jirovecii est nécessaire chez tous les patients pendant le traitement d’induction, quelle que soit la substance choisie (p. ex. Cotrim [Trimethoprim/Sulfamethoxazol] 480 mg 1×/jour). La prophylaxie peut être arrêtée une fois l’induction terminée et lorsque la dose de prednisolone <10-15 mg/j est atteinte. Le rôle de la plasmaphérèse reste controversé dans les formes graves telles que l’hémorragie alvéolaire diffuse (HAD). Bien que la société européenne de rhumatologie (EULAR) recommande de l’envisager dans sa dernière recommandation de 2016, l’étude PEXIVAS publiée dans le NEJM en février 2020, qui incluait 31 patients atteints d’HAD sévère dans le bras échange de plasma et 30 patients dans le bras de l’étude sans plasmaphérèse, n’a pas montré de bénéfice en termes de survie [2].
Une fois l’induction terminée, un traitement d’entretien doit être mis en place, le plus souvent avec du rituximab (2 administrations de 500 mg i.v. à 14 jours d’intervalle, puis 500 mg tous les 6 mois) ou alternativement avec de l’azathioprine (2 mg/Kg de poids corporel) pendant au moins 2 ans. Lors de l’initiation d’un traitement à l’azathioprine, des contrôles de laboratoire réguliers (hémogramme et transaminases) sont nécessaires et la co-médication avec des inhibiteurs de la xanthine oxydase (allopurinol et fébuxostat) est contre-indiquée. En outre, certains centres recommandent le dosage de la thiopurine S-méthyltransférase (TPMT) afin d’éviter une suppression sévère de la moelle osseuse chez les patients présentant le déficit enzymatique correspondant.
Les alternatives en cas de contre-indication/intolérance sont le méthotrexate (mais pas en cas d’atteinte rénale avec insuffisance rénale) ou le mycophénolate mofétil. Il convient toutefois de souligner que le mycophénolate mofétil n’est pas autorisé pour cette utilisation et qu’il est donc “off-label-use” et moins efficace que l’azathioprine sur la base des données de l’étude IMPROVE (hazard ratio pour la récidive sous mycophénolate mofétil vs. azathioprine 1,69) [3]. Le blocage du récepteur du complément C5a par l’avacopan constitue une nouvelle approche thérapeutique intéressante [4]. Dans l’étude présentée en ligne lors du Congrès européen de rhumatologie EULAR de cette année, cette substance administrée par voie orale a montré un effet comparable à celui du groupe témoin sous stéroïdes lorsqu’elle était utilisée en même temps que le traitement standard par cyclophosphamide ou rituximab, mais sans stéroïdes. L’Avacopan pourrait donc à l’avenir permettre de renoncer complètement aux stéroïdes dans ce groupe de maladies.
Granulomatose éosinophile avec polyangéite (EGPA)
L’EGPA représente un défi dans le groupe des vascularites associées aux ANCA. Il est difficile de confirmer le diagnostic, car l’histologie ne permet souvent pas de mettre en évidence une vascularite, les ANCA sont souvent négatifs et il faut exclure un syndrome hyperéosinophilique primaire comme expression d’une maladie clonale.
Une manifestation pulmonaire typique est l’apparition d’infiltrats fugaces. Histologiquement, la biopsie pulmonaire révèle souvent une éosinophilie tissulaire. De même, la décision de traitement n’est pas toujours facile à prendre en raison de l’insuffisance des études disponibles. En l’absence de paramètres pronostiques défavorables tels que l’atteinte rénale ou cardiaque, une monothérapie par corticostéroïdes est envisageable, sinon le cyclophosphamide est disponible pour les manifestations menaçant les organes ou le méthotrexate et l’azathioprine pour les évolutions moins sévères. Le rituximab n’a qu’une importance secondaire dans l’EGPA en raison de l’insuffisance des données disponibles. En revanche, l’utilisation de l’antagoniste de l’IL-5, le mépolizumab, qui a montré son efficacité dans une étude de phase 3 portant sur 136 patients atteints d’EGPA réfractaire ou récidivante, est intéressante.
Une administration s.c. 4 fois par semaine a permis de réduire la mortalité. L’administration de 300 mg de mépolizumab pendant 52 semaines a permis à davantage de patients d’obtenir une rémission (32% contre 3%) et de réduire la dose de prednisolone à 4 mg/j (44% contre 7%). Cependant, même dans le groupe mépolizumab, une rémission n’a pas été obtenue dans 47% des cas [5]. Entre-temps, le mépolizumab a également reçu l’approbation de la FDA pour l’EGPA.
Nœuds rhumatoïdes
Les nodules rhumatoïdes présentent une autre formation de granulomes. Ils sont surtout attendus chez les patients atteints de polyarthrite rhumatoïde séropositive (facteur rhumatoïde et/ou anticorps anti-CCP positifs) de longue date et de nodules rhumatoïdes cutanés. Il est difficile de les distinguer des tumeurs malignes sans histologie car, d’une part, il peut y avoir plusieurs nodules rhumatoïdes dont la taille augmente avec l’évolution et, d’autre part, les patients atteints de polyarthrite rhumatoïde présentent par se un risque plus élevé de tumeurs malignes. Typiquement, les nodules rhumatoïdes pulmonaires sont localisés souspleuraux ou dans la région des septa interlobulaires et sont généralement asymptomatiques (Fig. 3).
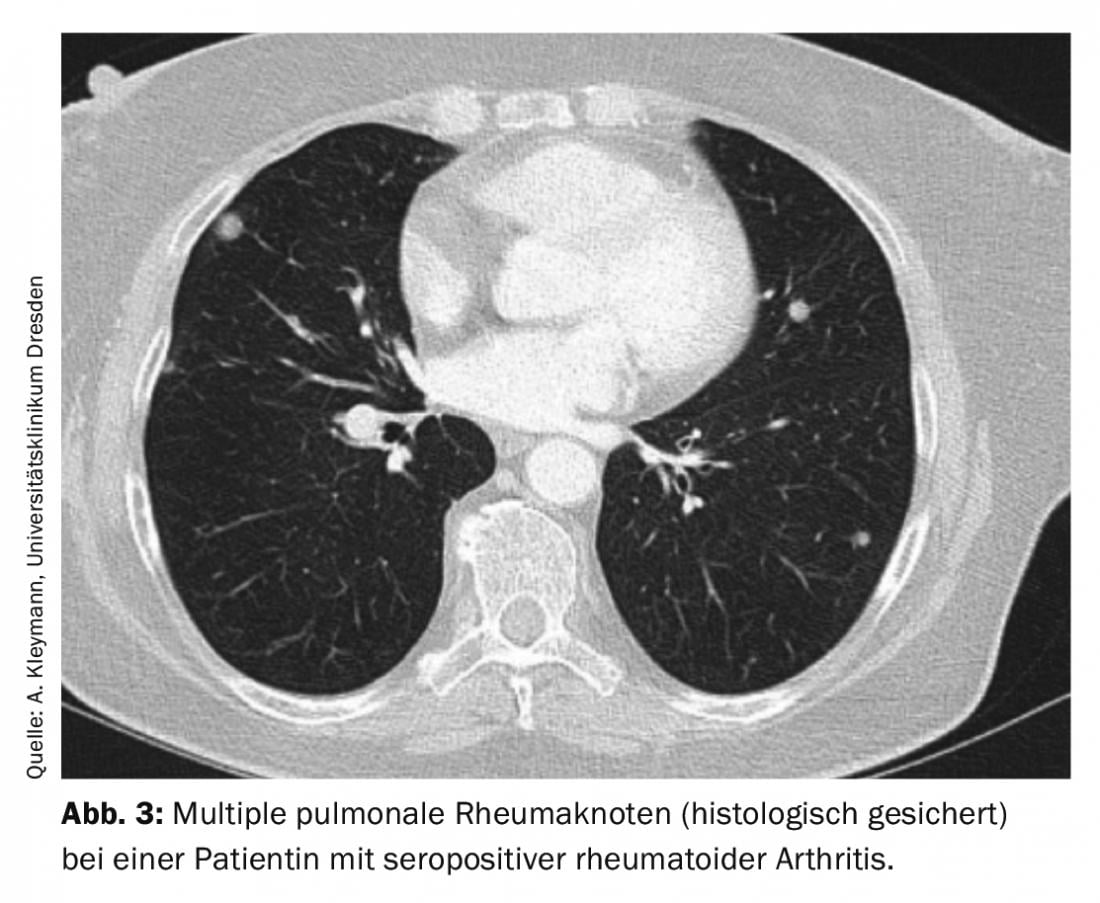
Cependant, ils peuvent également provoquer un épanchement pleural, un pneumothorax, une hémoptysie ou encore des fistules broncho-pulmonaires. En cas de détection de nodules rhumatoïdes, le traitement de base doit être adapté le cas échéant, car on observe plus souvent une augmentation de la taille des nodules rhumatoïdes sous méthotrexate. Des cas de développement de nodules rhumatoïdes pulmonaires ont également été décrits sous léflunomide [6,7].
Infections atypiques
En cas de réaction inflammatoire granulomateuse, il faut également envisager des infections atypiques sous le traitement de base plus immunosuppresseur. Sur le plan infectieux, il faut penser non seulement à la tuberculose, mais aussi aux infections fongiques telles que l’histoplasmose et la sporotrichose. Le risque d’infection chez les patients atteints de maladies rhumatismales inflammatoires est accru par la maladie de base, mais aussi par les traitements immunosuppresseurs de fond utilisés.
Avant tout , les bloqueurs du TNF semblent avoir une influence pertinente sur le risque d’infections pulmonaires granulomateuses. Cela est bien compris par l’importance physiopathologique du TNF-alpha dans la défense et la formation de granulomes contre les agents pathogènes bactériens et fongiques.
Histoplasmose
L’histoplasmose pulmonaire est causée par Histoplasma capsulatum. Cet agent pathogène ubiquitaire se trouve principalement en Amérique du Nord et en Amérique centrale. La distinction clinique avec la sarcoïdose et les tumeurs malignes est souvent difficile, car les manifestations pulmonaires sont variées (infiltrats pulmonaires ou foyers ronds, cavernes, lymphadénopathie médullaire ou masse) (Fig. 4).
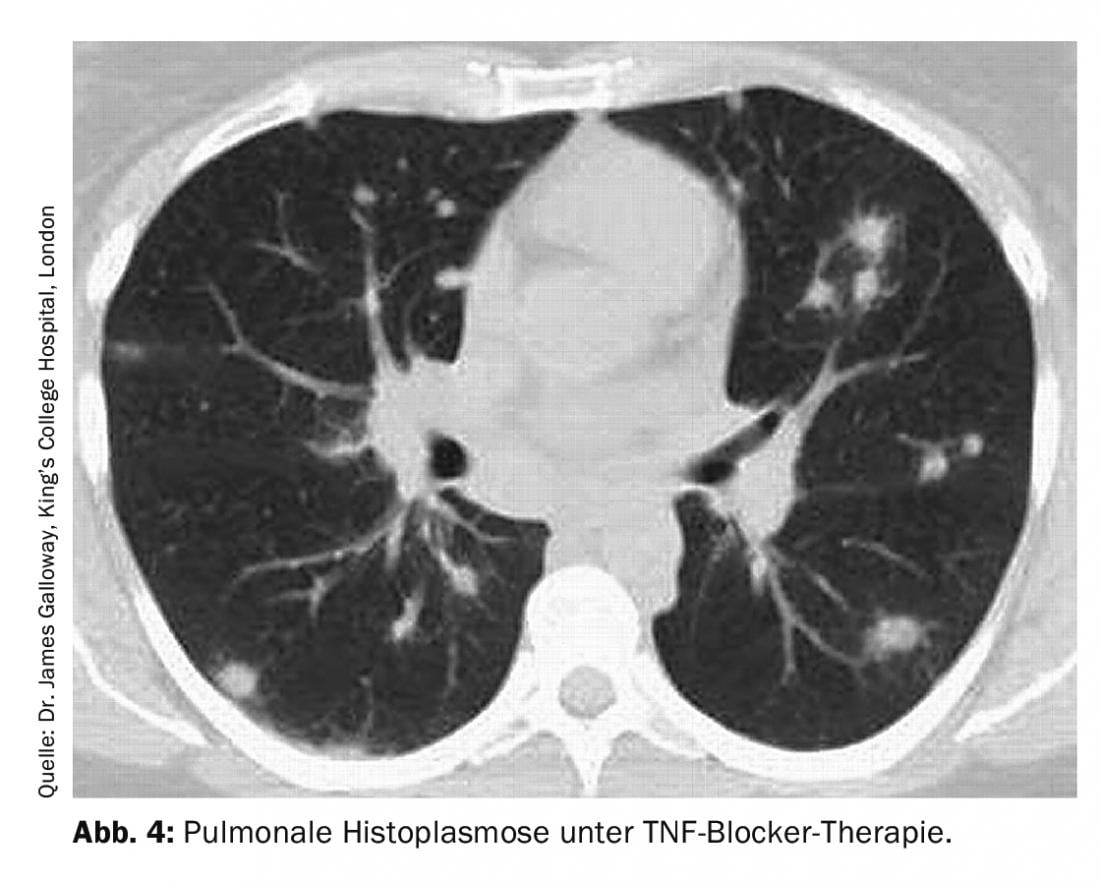
Le diagnostic repose sur la sérologie (anticorps spécifiques de l’histoplasme et détermination de l’antigène), mais aussi sur le lavage broncho-alvéolaire avec coloration fongique et culture, ainsi que sur l’examen histologique correspondant du biopsitogramme pour détecter les champignons. En fonction de la sévérité de la maladie, le traitement peut être soit absent, soit basé sur différents antifongiques.
Sporotrichose
La sporotrichose est causée par Sporothrix schenckii et a classiquement une manifestation cutanée. En cas de manifestation pulmonaire, la maladie ressemble à la tuberculose (figure 5). L’étalon-or du diagnostic est la détection culturelle de l’agent pathogène, car l’histopathologie peut être faussement négative en cas de faible nombre d’agents pathogènes, même avec une coloration appropriée, et les tests sérologiques et PCR ne sont actuellement pas encore disponibles en routine.
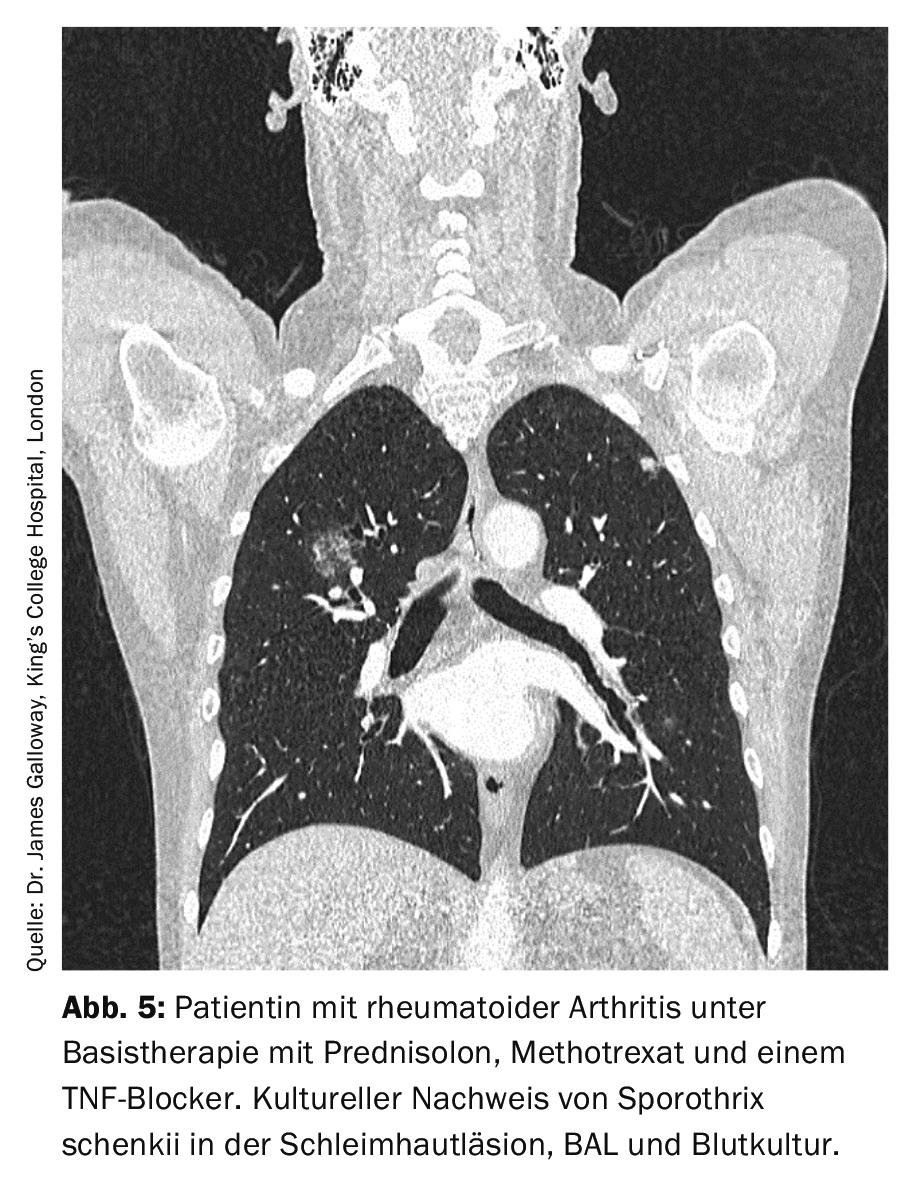
En l’absence de traitement, la sporotrichose pulmonaire est mortelle, de sorte qu’un traitement antifongique est toujours nécessaire. En cas d’évolution bénigne, il est possible d’administrer p.o. L’itraconazole 2×200 mg/d peut être utilisé. En cas de manifestation grave, il est d’abord recommandé d’administrer de l’amphotéricine B par voie i.v. et de ne passer à l’itraconazole qu’au fur et à mesure de l’évolution de la maladie. La durée du traitement doit être d’au moins un an [8].
Déficits immunitaires primaires
Enfin, il convient de mentionner le groupe complexe des déficits immunitaires primaires. En particulier, les patients présentant des infections récurrentes et des phénomènes auto-immuns doivent également penser à ce diagnostic différentiel. L’acronyme ELVIS décrit ici la susceptibilité pathologique aux infections (agents pathogènes et localisations atypiques, évolution prolongée, intensité et nombre d’infections exceptionnels [Summe]). L’acronyme GARFIELD résume ici les manifestations possibles comme expression d’une perturbation de la régulation immunitaire : Granulomes (figure 6), phénomènes auto-immuns, fièvre récurrente, lésions cutanées eczémateuses, lymphoprolifération (lymphadénopathie, splénomégalie) et inflammation chronique de l’intestin [9]. En cas de suspicion fondée, il convient d’initier un diagnostic plus approfondi avec un statut immunitaire cellulaire (hémogramme différentiel, typage des lymphocytes) et humoral (dosage des immunoglobulines, éventuellement avec des sous-classes d’IgG, facteurs du complément) et d’initier un examen approprié par un médecin expérimenté dans le diagnostic et le traitement des déficits immunitaires.
Messages Take-Home
- En présence de lésions pulmonaires granulomateuses, il faut également penser à des maladies rhumatologiques telles que la vascularite des petits vaisseaux, les nodules rhumatoïdes, mais aussi les infections atypiques sous traitement de fond et les déficits immunitaires primaires avec phénomène auto-immun.
- En cas de suspicion de vascularite pulmonaire, un diagnostic ANCA avec détermination des anticorps antiprotéinase 3 et myéloperoxydase doit être initié et, si le diagnostic est confirmé, une immunosuppression adéquate doit être mise en place rapidement.
- Les nodules rhumatoïdes pulmonaires ne sont pas rares en cas de polyarthrite rhumatoïde séropositive, mais il est souvent difficile de les distinguer des tumeurs malignes lors de la première manifestation.
- Les infections pulmonaires atypiques doivent être envisagées en particulier chez les patients traités par des médicaments biologiques et/ou des stéroïdes à fortes doses.
- Les déficits immunitaires primaires sont souvent associés à des phénomènes auto-immuns et doivent donc être exclus en cas de suspicion fondée.
Littérature :
- de Groot K, Harper L, Jayne DR, et al : Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cyto-plasmic antibody-associated vasculitis : a randomized trial. Ann Intern Med 2009 ; 150(10) : 670-680.
- Walsh M, et al : Échange de plasma et glucocorticoïdes dans les vascularites graves associées aux ANCA. N Engl J Med 2020 ; 382 : 622-631.
- Hiemstra TF, Walsh M, Mahr A, et al : Mycophénolate mofetil vs azathioprine pour le maintien de la rémission dans la vascularite associée à un anticorps cytoplasmique antineutrophile : un essai contrôlé randomisé. JAMA 2010 ; 304(21) : 2381-2388.
- Merkel PA, Jayne DR, Wang C, et al. : Évaluation de la sécurité et de l’efficacité d’Avacopan, un inhibiteur du récepteur C5a, chez les patients atteints de vascularite associée à un anticorps cytoplasmique antineutrophile et traités de manière concomitante par rituximab ou cyclophosphamide/azathioprine : Protocole pour un essai de phase 3 randomisé, en double aveugle et contrôlé de manière active. JMIR Res Protoc 2020 ; 9(4) : e16664.
- Wechsler ME, Akuthota P, Jayne D, et al : Mepolizumab or Placebo for Eosinophilic Granulomatosis with Polyangiitis. N Engl J Med 2017 ; 376(20) : 1921-1932.
- Horvath IF, Szanto A, Csiki Z, et al : Nodules rhumatoïdes intrapulmonaires chez un patient souffrant d’arthrite rhumatoïde de longue date et traité au leflunomide. Pathol Oncol Res 2008 ; 14(1) : 101-104.
- Rozin A, Yigla M, Guralnik L, et al : Rhumatoid lung nodulosis and osteopathy associated with leflunomide therapy. Clin Rheumatol 2006 ; 25(3) : 384-388.
- Kauffman CA, Bustamante B, Chapman SW, et al : Clinical practice guidelines for the management of sporotrichosis : 2007 update by the Infectious Diseases Society of America. Clin Infect Dis 2007 ; 45(10) : 1255-1265.
- Ligne directrice de l’AWMF “Diagnostic de la présence d’une immunodéficience primaire”, version du 31 octobre 2017, valable jusqu’au 31 octobre 2020.
InFo PNEUMOLOGIE & ALLERGOLOGIE 2020 ; 2(3) : 12-16