Les maladies de stockage lysosomales sont un groupe hétérogène de maladies génétiques qui trouvent leur origine dans un dysfonctionnement des processus métaboliques lysosomaux. La maladie de Niemann-Pick est aujourd’hui également appelée ASMD (“Acid Sphingomeyelinase Deficiency”). Les types A et B sont classés parmi les sphingolipidoses, tandis que le type C fait partie des maladies de stockage des lipides. Pour les types A et B, le premier traitement de substitution enzymatique a été approuvé dans l’UE l’année dernière.
La maladie de Niemann-Pick est une maladie génétique de stockage lysosomale qui doit son nom au pédiatre allemand Albert Niemann (1880-1921) et au pathologiste allemand Ludwig Pick (1868-1944). Ludwig Pick a réussi à distinguer la maladie de Niemann-Pick de la maladie de Gaucher en tant que maladie métabolique distincte [1]. La substance de stockage, la sphingomyéline, a été mise en évidence par le biochimiste Klenk en 1934. Une classification en différents sous-groupes a été initiée par Crocker en 1961 [2–4]. Les formes de manifestation les plus courantes sont de type A-C (encadré).
| Les sous-formes les plus courantes Le syndrome de Niemann-Pick est une maladie héréditaire autosomique récessive. Les types A et B sont dus à une activité déficiente d’une enzyme lysosomale codée par le gène SMPD1. Le défaut génétique fait que la sphingomyéline ne peut plus être dégradée et s’accumule dans les cellules de différents organes. Le type C est un trouble du métabolisme du cholestérol dans lequel des mutations du gène NPC-1 (18q11) ou du gène NPC-2 (14q24.3) sont détectables. |
| vers [11] |
Manifestations cliniques
Le type A est une maladie neurodégénérative grave du nourrisson, qui entraîne généralement la mort au cours des trois premières années de vie. Les principaux symptômes sont une hépatosplénomégalie et une dégradation psychomotrice. Le développement des enfants concernés stagne. Les compétences acquises au cours des années précédentes se perdent avec le temps. Avant la fin du premier semestre de vie, on observe souvent des troubles de la croissance, des vomissements, une baisse de l’audition, une tétraspasticité et des crises myocloniques.
Le type B se caractérise par un début plus tardif de la maladie et une manifestation plus légère que le type A [5]. La plupart des patients atteignent l’âge adulte et aucun symptôme cérébral n’est observé. En revanche, une hépatosplénomégalie avec hypersplénisme progressif et un dysfonctionnement hépatique stable, ainsi qu’une détérioration progressive de la fonction pulmonaire, accompagnée d’une ostéopénie et d’un profil lipidique athérogène, sont caractéristiques [6]. Un profil lipidique proathérogène est observé très tôt dans l’évolution de la maladie, et certains patients développent une maladie coronarienne.
Le type C est associé à une altération du transport du cholestérol à partir des lysosomes, ce qui entraîne une augmentation du stockage du cholestérol, des gycosphingolipides et des gangliosides dans les lysosomes de différentes cellules de l’organisme [7,8]. Par rapport au type A/B, l’aspect clinique est très hétérogène. Il s’agit d’une maladie neuroviscérale chronique, dont la progression est plus lente que celle du type A. Elle se caractérise par une perte d’appétit et des troubles du sommeil. On peut la diviser en une forme infantile précoce, une forme infantile tardive, une forme juvénile et une forme adulte [7]. Cliniquement, les personnes atteintes présentent diverses anomalies neurologiques et psychiatriques, parfois des symptômes viscéraux comme une hépatosplénomégalie [8]. Les manifestations neurologiques typiques du type C sont des troubles cognitifs, des crises d’épilepsie, des troubles du comportement, une dépression et des psychoses, une parésie verticale du regard, des troubles de la parole et de la déglutition et des dystonies [9].
| Type A/B : nouvelle thérapie de substitution enzymatique : Olipudase alfa Pour les patients pédiatriques et adultes atteints de la maladie de Niemann-Pick ou d’ASMD (“acid sphingomeyelinase deficiency”) de type A/B ou de type B sans atteinte du système nerveux central, l’EMA a autorisé en 2022 le traitement enzymatique substitutif par l’olipudase alfa. L’olipudase alfa est destinée à remplacer l’ASM manquante ou défectueuse afin de permettre la dégradation de la sphingomyéline. La décision d’autorisation de mise sur le marché est basée sur les données des études cliniques ASCEND et ASCEND-Peds, qui ont montré des améliorations cliniquement significatives de la fonction pulmonaire et une réduction du volume de la rate et du foie sous traitement par l’olipudase alfa. La fréquence des événements indésirables était comparable chez les patients recevant l’olipudase alfa et dans le groupe placebo. L’olipudase alfa est perfusée toutes les deux semaines pendant la phase d’entretien. Dans l’étude ASCEND, 36 patients adultes atteints d’ASMD de type A/B ou de type B ont été randomisés pour recevoir de l’olipudase alfa ou un placebo. Après 52 semaines, le bras de traitement a montré une amélioration de la fonction pulmonaire et une réduction du volume de la rate. Dans l’étude ASCEND-PEDS à un bras, 20 patients pédiatriques atteints d’ASMD de type A/B ou de type B ont été traités par l’olipudase alfa pendant 64 semaines. Ici aussi, les principaux critères d’évaluation ont été atteints au cours de la semaine 52. |
| vers [12] |
Diagnostic et traitement
Les cultures de leucocytes et de fibroblastes permettent de mettre en évidence une activité réduite ou absente de la sphingomyélinase acide. La cause de la maladie de Niemann-Pick de type A et B est [10]. Ces examens enzymatiques et de génétique moléculaire peuvent être effectués dès la période prénatale si l’on connaît des antécédents familiaux [1]. Pour établir le diagnostic de la maladie de Niemann-Pick de type C , il faut procéder à des examens complexes du métabolisme du cholestérol [10]. Les anomalies génétiques sous-jacentes ne peuvent actuellement pas être traitées (situation en 2022). Pour les types A et B de la maladie de Niemann-Pick, un traitement enzymatique substitutif par l’olipudase alfa est disponible dans l’UE (encadré). Le type C est traité de manière symptomatique avec du miglustat.
Littérature :
- “Examen du foie et de la rate par élastographie chez des patients atteints de la maladie de Niemann-Pick de type B”, Gözde Aksu, thèse inaugurale, 2020, https://openscience.ub.uni-mainz.de,(dernière consultation 12.10.2023)
- Crocker AC, Mays VB : Synthèse de la sphingomyéline dans la maladie de Niemann-Pick. Am J Clin Nutr 1961 ; 9 : 63-67.
- Crocker AC : Le déficit cérébral dans la maladie de Tay-Sachs et la maladie de Niemann-Pick. J Neurochem 1961 ; 7 : 69-80.
- E. K. Sur la nature des phosphatides de la rate dans la maladie de Niemann-Picksen. Revue de chimie physiologique de Hoppe Seyler. 1934.
- Tran C, et al. : Implication pulmonaire chez les patients adultes atteints de maladies métaboliques congénitales. Karger Kompass Pneumol 2018 ; 6 : 6-17.
- Orphanet, www.orpha.net,(dernière consultation 12.10.2023)
- Di Lazzaro V, et al : Niemann-Pick type C : focus sur la forme d’onset adolescent/adulte. Int J Neurosci 2016 ; 126(11) : 963-971.
- Hammerschmidt TG, et al : Marqueurs biologiques moléculaires et biochimiques pour le diagnostic et le suivi thérapeutique des patients Niemann-Pick type C. Int J Dev Neurosci 2017 ; 66 : 18-23.
- Bonnot O, et al. : Symptômes psychiatriques et neurologiques chez les patients atteints de la maladie de Niemann-Pick de type C (NP-C) : Findings from the International NPC Registry. World J Biol Psychiatry 2017 : 1-10.
- “Morbus Niemann-Pick”, https://flexikon.doccheck.com,(dernière consultation 12.10.2023)
- Desnick JP, et al : Identification et caractérisation de huit nouvelles mutations SMPD1 causant les types A et B de la maladie de Niemann Pick. Mol Med 2010 ; 16 : 316-321.
- “Xenpozyme® (olipudase alfa) approuvé par la Commission européenne comme premier et seul traitement de l’ASMD”, 28.06.2022.
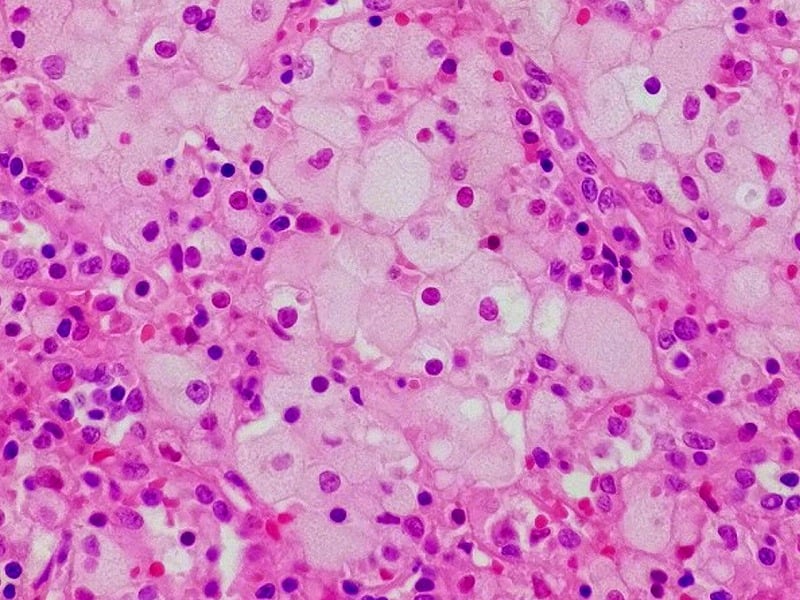
| Image de couverture : Niemann pick cell in spleen. W.CC, Wikimedia |
PRATIQUE DU MÉDECIN DE FAMILLE 2023 ; 18(10) : 48