
L’espérance et la qualité de vie des personnes atteintes de SLA peuvent être améliorées grâce à un traitement moderne. La volonté du patient est primordiale et doit être déterminée à chaque fois. Il n’y a pas de nouveautés en matière de traitement médicamenteux – le riluzole doit être commencé tôt. Au début de la maladie, il est utile de procéder à une évaluation détaillée dans le cadre hospitalier d’une clinique neurologique. La poursuite du traitement dans des centres spécialisés au cours de l’évolution de la maladie est recommandée.
La sclérose latérale amyotrophique (SLA), qui est la maladie du motoneurone la plus fréquente, se caractérise par la destruction progressive des cellules nerveuses du système moteur. Les premiers motoneurones de la voie pyramidale et les deuxièmes motoneurones des cellules de la corne antérieure sont typiquement concernés. Les signes du premier ou du deuxième motoneurone peuvent être prédominants. L’éventail des maladies du motoneurone comprend d’autres maladies telles que la sclérose latérale primaire (SLA) ou l’amyotrophie spinale (SMA), qui ne touchent respectivement que le premier ou le deuxième nerf moteur. deuxième motoneurone.
L’incidence de la SLA est rare par rapport à d’autres maladies. L’incidence est tout de même d’environ 2/100 000 habitants, ce qui n’est que légèrement inférieur à l’incidence de la sclérose en plaques, par exemple. En revanche, la prévalence est très faible, de 3 à 8/100 000 habitants [1]. Cela reflète indirectement la courte survie moyenne des patients après le diagnostic, qui n’est que de deux à quatre ans dans la majorité des cas. Il est toutefois intéressant de noter qu’environ 10% des patients ont une évolution beaucoup plus lente, avec une survie supérieure à dix ans.
Grâce à un traitement de soutien moderne et maximal, il est désormais possible de prolonger considérablement la survie des malades et d’améliorer leur qualité de vie, du moins en ce qui concerne les symptômes essentiels tels que la douleur, la faim et la détresse respiratoire. Mais à la lumière de l’amélioration des possibilités médicales, il est très important de toujours placer le souhait de la personne concernée au centre des décisions thérapeutiques [2]. Pour cela, il convient de rédiger très tôt des directives anticipées détaillées, qui devront être revues au fur et à mesure de l’évolution de la maladie.
Principes de diagnostic
Les pierres angulaires du diagnostic de la SLA restent l’anamnèse et l’examen clinique. Lorsqu’elles sont effectuées par un neurologue expérimenté, elles fournissent généralement déjà des indications claires sur la présence d’une lésion progressive du premier et/ou du deuxième motoneurone. Ce seul élément permet de diagnostiquer une SLA probable ou certaine selon les critères de diagnostic en vigueur [3]. Les résultats cliniques peuvent être étayés par des résultats électrophysiologiques, qui sont désormais de plus en plus pris en compte dans l’algorithme de diagnostic (Fig. 1) [4].
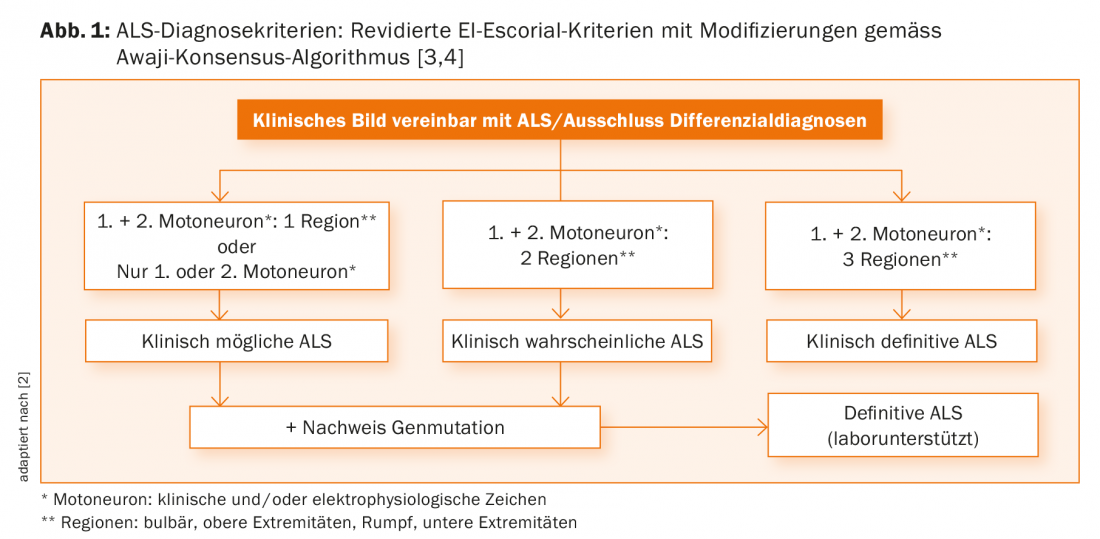
Malheureusement, les critères actuels ne permettent souvent de poser un diagnostic sûr que tardivement dans l’évolution de la maladie, ce qui peut, d’une part, inquiéter les prescripteurs, les patients et leurs proches et, d’autre part, ne constitue pas une bonne base pour des études scientifiques. C’est pourquoi la révision des critères de diagnostic reste d’une grande importance.
Une tâche diagnostique essentielle consiste à exclure tout diagnostic différentiel susceptible de provoquer une constellation de symptômes comparable. Celles-ci sont clairement résumées dans les lignes directrices actuelles de l’EFNS [2]. A titre d’exemple, l’association pas si rare d’une sténose du canal rachidien cervical et d’une polyneuropathie peut tout à fait fournir la combinaison de signes du premier et du deuxième motoneurone nécessaire au diagnostic de la SLA. Une évaluation hospitalière dans une clinique neurologique au début de la maladie a fait ses preuves, afin de disposer de suffisamment d’espace et de temps pour l’évaluation clinique, l’exclusion des diagnostics différentiels et la communication empathique du diagnostic.
Examen clinique
L’examen clinique vise d’abord à rechercher des signes d’atteinte du premier et du deuxième motoneurone dans les quatre régions du corps (bulbaire, membres supérieurs, tronc, membres inférieurs). Les signes du premier motoneurone comprennent la spasticité, le clonisme, l’hyperréflexie et les phénomènes de désinhibition centrale. Les signes cliniques du deuxième motoneurone sont principalement des fasciculations et des atrophies. En outre, il convient de rechercher des signes atypiques lors de l’examen clinique.
Par exemple, l’implication des muscles oculaires ou l’absence de progression orientent le regard vers d’autres diagnostics. En revanche, les troubles de la sensibilité n’excluent pas complètement une SLA, mais ils doivent certainement eux aussi donner lieu à un examen approfondi des diagnostics différentiels.
Électrophysiologie
L’électroneurographie (ENG) permet d’exclure une polyneuropathie. À l’électromyographie (EMG), une constellation de symptômes comprenant des signes de lésions aiguës, subaiguës et éventuellement chroniques est typique de la SLA, reflétant l’évolution de la maladie et la capacité préservée des axones périphériques à se régénérer. Outre la recherche d’une activité spontanée pathologique sur le muscle au repos, une attention particulière est accordée à la stabilité et à la taille des unités motrices dans l’analyse du potentiel individuel. Un résultat pathologique approprié à l’EMG est un élément essentiel du diagnostic de la SLA et peut être considéré comme un signe clinique selon les critères de diagnostic modifiés [4].
Les potentiels évoqués moteurs peuvent détecter des lésions subcliniques de la voie pyramidale. Les méthodes de quantification des unités motrices (estimation du nombre d’unités motrices) sont des méthodes encore plus scientifiques. Ces méthodes pourraient revêtir une importance croissante, notamment pour l’évolution de la maladie et donc pour les études cliniques [5].
Imagerie
La place de l’échographie dans le diagnostic de la SLA augmente depuis quelques années, même s’il est peu probable que cette méthode atteigne l’importance qu’elle a dans les maladies nerveuses périphériques. L’échographie nerveuse peut surtout aider à exclure des diagnostics différentiels pertinents tels que les neuropathies à médiation immunitaire. Les ultrasons permettent également d’examiner les muscles. Pour la détection des fasciculations, l’échographie musculaire, avec une excellente sensibilité mais une spécificité un peu plus faible, pourra peut-être à l’avenir remplacer, du moins en partie, l’électromyographie, notamment pour les examens d’évolution [6]. L’IRM est surtout importante pour exclure les diagnostics différentiels. Dans la plupart des cas, le cerveau et l’ensemble de la colonne vertébrale sont représentés. En outre, les muscles et les nerfs périphériques peuvent également être visualisés, bien que l’accent soit encore une fois mis sur le domaine scientifique plutôt que sur la pratique clinique.
se trouve.
Diagnostic génétique
Le diagnostic génétique de la SLA s’est enrichi de plusieurs aspects au cours des dernières années. Pendant de nombreuses années, on a recherché en particulier des mutations dans le gène de la superoxyde dismutase SOD1 de Cu/Zn, qui sont responsables d’environ 10 à 15% des cas familiaux de SLA. Paradoxalement, il existe des familles dans lesquelles la détection de la mutation n’est pas corrélée à l’apparition de la maladie, ce qui indique qu’il doit exister d’autres facteurs ou gènes responsables de la maladie [7]. Entre-temps, un nouveau locus important a été trouvé : le gène C9orf72. Des mutations dans ce gène sont responsables d’environ 25% des cas familiaux et d’environ 10% des cas sporadiques. Il est intéressant de noter que des mutations dans ce gène existent également dans une proportion similaire de cas de démence fronto-temporale, ce qui illustre le lien entre les deux maladies, y compris au niveau génétique [8].
Des tests génétiques des mutations les plus courantes sont aujourd’hui disponibles dans le commerce. Il est d’autant plus important de souligner que le diagnostic génétique doit être confié à des mains expérimentées, en particulier dans le cas de la SLA. Elle ne devrait être demandée qu’après un examen approfondi et des conseils de neurologues et de généticiens humains familiarisés avec la maladie, car le résultat peut, dans certaines circonstances, avoir une grande pertinence, notamment pour les conseils aux membres de la famille asymptomatiques.
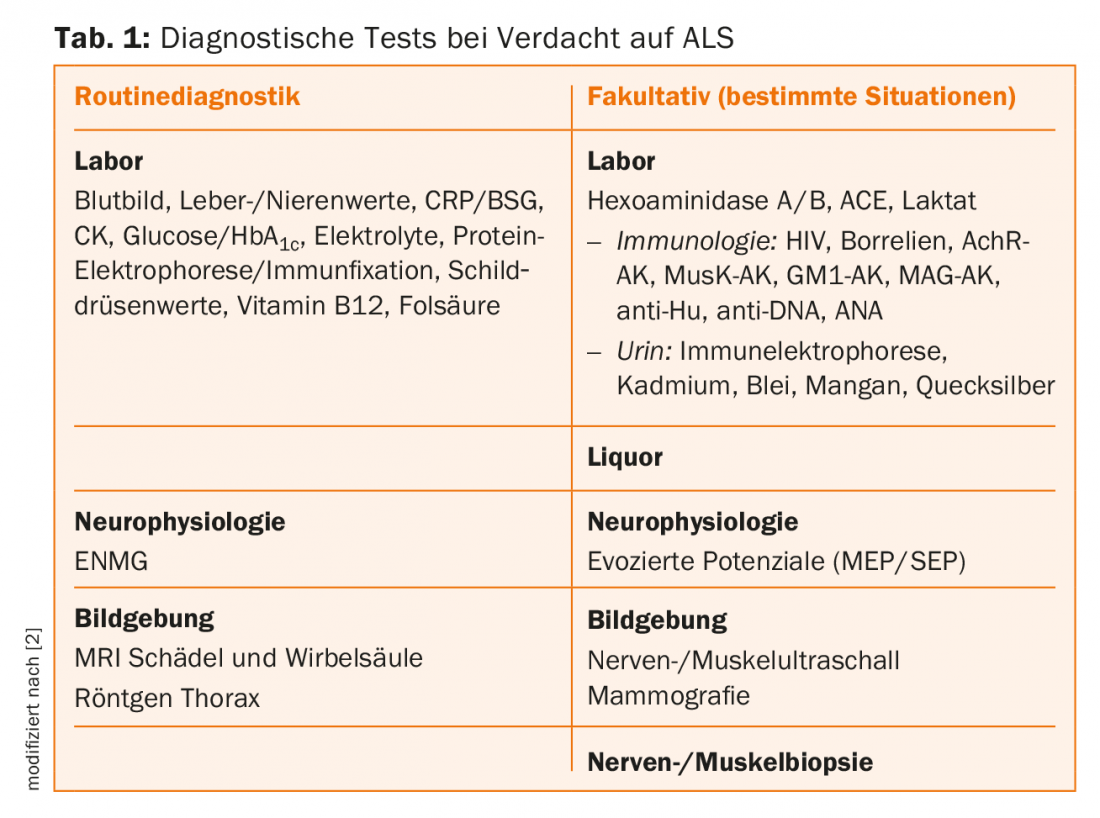
Diagnostics supplémentaires
En cas de suspicion de troubles cognitifs, un test neuropsychologique est recommandé. Selon la durée de la maladie, jusqu’à 50% des patients atteints de SLA développent des symptômes dysexécutifs, et chez environ 15% d’entre eux, un diagnostic de démence fronto-temporale peut être posé. Cette observation va à l’encontre des doctrines antérieures qui ne voyaient pas d’altération cognitive dans la SLA. Par ailleurs, les patients atteints de démence fronto-temporale doivent également faire l’objet d’une recherche approfondie des signes d’une maladie du motoneurone, qui est également fréquente au cours de l’évolution de la maladie. Sinon, le reste du diagnostic sert essentiellement à exclure les diagnostics différentiels [2].
Thérapie médicamenteuse
De plus, le riluzole est le seul médicament autorisé pour le traitement de la SLA et il est recommandé de le prescrire le plus tôt possible dans l’évolution de la maladie. Le médicament est sans danger et, outre une légère fatigue, on observe rarement une augmentation des enzymes hépatiques. Le riluzole permet de ralentir quelque peu l’évolution de la maladie. En outre, d’autres traitements médicamenteux peuvent être utilisés en fonction des symptômes, comme par exemple l’amitriptyline ou des gouttes d’atropine en cas de salivation gênante. Les médicaments potentiellement sédatifs tels que les benzodiazépines et les opiacés peuvent être très bien utilisés pour traiter l’anxiété, la douleur et la dyspnée, en tenant compte de limites thérapeutiques relativement étroites.
Nutrition
La perte de poids est un marqueur pronostique négatif qui doit être évité autant que possible. Les malades et leur famille peuvent recevoir des conseils appropriés et des suppléments alimentaires peuvent être offerts. Un suivi orthophonique rapproché pour détecter précocement une dysphagie est essentiel pour que la proposition d’une alimentation entérale percutanée par sonde PEG puisse être faite le plus tôt possible. Plus la pose d’un PEG est précoce dans l’évolution de la maladie, plus le risque de complications est faible et plus cette mesure peut contribuer à améliorer la qualité de vie et la durée de survie. Ce qui est important pour les personnes concernées, c’est qu’une alimentation orale supplémentaire reste bien entendu possible.
Respiration artificielle
A tout moment de l’évolution de la maladie, il faut être attentif aux symptômes d’hypoventilation, surtout nocturnes. Outre la dyspnée, il s’agit de la somnolence diurne, des maux de tête matinaux ou de l’orthopnée. Ces symptômes peuvent être traités très efficacement par une ventilation non invasive à domicile, ce qui améliore considérablement la qualité de vie des personnes concernées. Ce traitement symptomatique efficace devrait être abordé rapidement avec toutes les personnes concernées dès l’apparition des symptômes correspondants, car il peut, comme peu d’autres traitements, améliorer la qualité de vie des personnes concernées, du moins pendant une période limitée [9]. En revanche, la ventilation invasive n’est envisageable que pour un groupe de patients sélectionnés. Si, à un moment donné de l’évolution de la maladie, le patient souhaite arrêter la ventilation, il faut respecter ce souhait et l’accompagner en conséquence. En général, cette mesure entraîne rapidement unerétention de CO2, les patients décédant dans uneanesthésie au CO2 qui s’installe lentement.
Autre thérapie
Un traitement spécialisé et multiprofessionnel étroit, tel qu’il est dispensé dans un centre SLA ou une institution similaire, est essentiel pour une prise en charge optimale des personnes atteintes de SLA. D’autre part, d’autres mesures thérapeutiques telles que la physiothérapie, l’ergothérapie et l’orthophonie peuvent être complétées de manière judicieuse afin de préserver le plus longtemps possible les fonctions encore préservées, comme par exemple la capacité de communication.
Littérature :
- Schweikert K : Sclérose latérale amyotrophique. Swiss Medical Forum 2015 ; 15 : 1068-1073.
- Andersen PM, et al : Lignes directrices de l’EFNS sur la prise en charge clinique de la sclérose latérale amyotrophique (SLA) – rapport révisé d’un groupe de travail de l’EFNS. Eur J Neurol 2012 ; 19 : 360-375.
- Brooks BR, et al : El Escorial revisité : critères révisés pour le diagnostic de la sclérose latérale amyotrophique. Amyotroph Lateral Scler Other Motor Neuron Disord 2000 ; 1 : 293-299.
- Carvalho MD, et al : Awaji algorithm diagnostic increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler 2009 ; 10 : 53-57.
- Schulte-Mattler WJ, et al : MUNIX – un biomarqueur prometteur dans la SLA. Neurophysiol clinique 2015 ; 46 : 186-189.
- Schreiber S, et al. : Expérience et place des méthodes d’imagerie dans les changements neuromusculaires induits par la SLA. Neurophysiol clinique 2016 ; 46 : 173-181.
- Felbecker A, et al. : Quatre pedigrees familiaux SLA discordants pour deux mutations SOD1 : toutes les mutations SOD1 sont-elles pathogènes ? J Neurol Neurosurg Psychiatry 2010 ; 81 : 572-577.
- Rohrer JD, et al : C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 2015 ; 14 : 291-301.
- Boentert M, et al : Ventilation et gestion des sécrétions dans la sclérose latérale amyotrophique. Neurophysiol clinique 2015 ; 46 : 163-172.
InFo NEUROLOGIE & PSYCHIATRIE 2016 ; 14(2) : 26-29









