Les tumeurs neuroendocrines (TNE) ne sont pas si rares. La prévalence de ces tumeurs dans le tractus gastro-intestinal n’est dépassée que par le cancer du côlon. Souvent, les NET ne sont détectées qu’à un stade tardif en raison de la subtilité des symptômes et sont souvent déjà à un stade palliatif et inopérable. Il est donc important de penser à ces tumeurs. De nombreuses nouvelles possibilités de traitement chirurgical, interventionnel local et systémique ont été introduites au cours des dernières années. En raison du degré élevé de complexité du diagnostic et du traitement de ces tumeurs, il est souhaitable de travailler au sein d’une équipe spécialisée composée de gastro-entérologues, de chirurgiens, de pathologistes, de radiologues, d’experts en médecine nucléaire, d’endocrinologues et d’oncologues.
Les tumeurs neuroendocrines (TNE) ont une incidence relativement faible, mais croissante, de 4 à 5 nouveaux cas / 100 000 habitants par an [1]. Cependant, la prévalence, c’est-à-dire le nombre de patients atteints de tumeurs neuroendocrines gastro-intestinales (TNE-GEP), est bien plus élevée et n’est dépassée dans le tractus gastro-intestinal que par le cancer du côlon. De plus, l’incidence de toutes les GEP-NET augmente, à l’exception des carcinoïdes appendiculaires [2]. Dans la pratique, les GEP-NET sont plus fréquents que prévu. Ce bref aperçu résume les faits les plus importants concernant le diagnostic, les différentes formes de diagnostic de propagation, les principales mesures thérapeutiques ainsi que le suivi. Ce résumé s’appuie sur les directives actuelles de la Société européenne des tumeurs neuroendocrines(www.enets.org).
Classification des GEP-NET
Les tumeurs neuroendocrines sont principalement classées en fonction de la localisation de la tumeur primaire et du nombre de mitoses (Ki67). Le taux de mitoses est l’un des facteurs pronostiques les plus importants et classe ces tumeurs en G1 (Ki67 ≤ 2%), G2 (Ki67 3-20%) et G3 (Ki67 >20%), le stade G3 correspondant à un carcinome neuroendocrine moins différencié et souvent agressif par rapport aux tumeurs neuroendocrines mieux différenciées [3,4].
Les tumeurs neuroendocrines gastro-intestinales peuvent apparaître à n’importe quel endroit du tube digestif. Les caractéristiques et la forme des métastases varient en fonction de la localisation de la tumeur primaire et nécessitent des investigations différentes en conséquence. L’incidence des NET gastriques (g-NET) n’a cessé d’augmenter au cours des dernières années et représente environ 8% des NET GEP [5]. Les raisons en sont certainement l’introduction généralisée de la gastroscopie et peut-être l’utilisation fréquente d’inhibiteurs de la pompe à protons [6].
Les g-NET sont divisés en trois sous-groupes, le type I, le plus fréquent, pouvant être associé à une gastrite atrophique, étant généralement bien différencié, rarement métastatique et de bon pronostic. Les sous-types II et III sont plus rares, plus souvent métastatiques et ont un moins bon pronostic (surtout le sous-type III) [5]. L’évaluation et le pronostic de ces tumeurs gastriques doivent être adaptés en fonction de leur type et de leur taille. Les NET duodénales (d-NET) sont rares (1- 2% des NET GEP), apparaissent généralement dans les deux premiers tiers du duodénum, sont normalement relativement petites, plutôt bien différenciées et métastasent néanmoins localement dans 40-60% dans les ganglions lymphatiques [7]. L’incidence des GEP-NET pancréatiques (p-NET) se situe entre 2 et 3/100 000 personnes par an [8]. Dans le p-NET, les signes précoces d’une maladie maligne sont souvent absents. Ces patients se présentent chez leur médecin en cas de compression locale ou de signes de métastases avancées (notamment au niveau du foie). Pour cette raison, plus de 50% des patients atteints de p-NET présentent déjà des métastases à distance.
La néoplasie endocrinienne multiple de type I (MEN I) est une maladie sous-jacente globalement rare mais typique des p-NET (également des d-NET).
Environ 20% des patients atteints de MEN1 souffrent d’un p-NET, avec une incidence croissante avec l’âge [8]. De même, les patients atteints de la maladie de Von Hippel-Lindau, entre autres dégénérescences malignes, ont une probabilité plus élevée de développer un p-NET.
Les TNE jéjunales et iléales sont les plus fréquentes des TNE GEP, représentant jusqu’à 30% de tous les cas [9]. Les TNE de l’intestin grêle, comme les TNE p, ont tendance à être déjà métastasées au moment du diagnostic. En général, les ganglions lymphatiques locaux et, plus tard, le foie sont souvent touchés. Les TNE du côlon représentent environ 7% de toutes les TNE GEP et ont généralement le plus mauvais pronostic [10]. Comme les tumeurs décrites précédemment, les TNE du côlon sont généralement très avancées au moment du diagnostic et métastasent localement, dans le péritoine et dans le foie. En revanche, les NET rectale sont souvent détectées relativement tôt. Ceci en raison de symptômes précoces, tels que des douleurs ou du sang abo. Pour cette raison, entre autres, les métastases de la TNE rectale sont plutôt rares [10]. Dans l’ensemble, la biologie des GEP-NET varie en fonction de leur localisation et de leur différenciation. C’est pourquoi le diagnostic, le traitement et le suivi doivent être adaptés à la localisation primaire.
Évaluation diagnostique et imagerie
L’évaluation de la GEP-NET diffère selon la localisation. Le diagnostic est établi par une confirmation histologique [11]. Une ponction à l’aiguille fine pour un traitement cytologique est souvent insuffisante. Les marqueurs obligatoires pour établir le diagnostic sont la synaptophysine et la chromogranine A. De même, le degré de différenciation (degré G1-G3) doit impérativement être relevé. D’autres marqueurs, par exemple les récepteurs individuels de la somatostatine, ne sont pas obligatoires, mais sont intéressants, par exemple pour les étapes thérapeutiques ultérieures.
Une fois le diagnostic posé, on procède au diagnostic d’extension (staging) [12]. Le staging est, avec le grading, le marqueur pronostique le plus important pour le patient. Les méthodes utilisées comprennent l’échographie (y compris l’échographie endoscopique), la TDM souvent avec contraste, l’IRM et les méthodes fonctionnelles telles que la scintigraphie à l’octréotide et la TEP/TDM au gallium 68 DOTATATE/DOTATOC et la TEP/TDM au fluor-18 désoxyglucose (FDG). Le domaine de l’échographie est surtout la visualisation des métastases hépatiques, avec une sensibilité et une spécificité de l’ordre de 90%. L’échographie endoscopique (EUS) est particulièrement utilisée pour les tumeurs neuroendocrines pancréatiques (par exemple, les insulinomes pancréatiques ou les tumeurs pancréatiques non fonctionnelles). La spécificité/sensibilité est supérieure à 90%. Les tumeurs duodénales sont plus difficiles à visualiser par EUS. Le taux de détection de ces tumeurs par EUS n’est plus que de 60%. La TDM avec contraste est la méthode la plus répandue pour la stadification des GEP-NET. Il est important de réaliser une phase artérielle, notamment en cas de métastases hépatiques. Cela augmente le taux de détection des métastases hépatiques à environ 80%. Cependant, la sensibilité est légèrement inférieure (autour de 70%), surtout pour le p-NET et les métastases des tissus mous. Ces tumeurs sont le domaine typique de l’examen IRM. L’IRM a une sensibilité de plus de 90% pour les p-NET dans le pancréas. En outre, l’IRM hépatique avec produit de contraste spécifique au foie est l’examen de choix pour détecter les métastases hépatiques. Avec un taux de détection pouvant atteindre 95%, l’IRM hépatique est la méthode la plus sensible pour les métastases hépatiques. Les méthodes fonctionnelles sont typiquement utilisées pour le diagnostic de propagation et pour mesurer l’expression des récepteurs de la somatostatine (en particulier SSTR2) [13].
La scintigraphie à l’octréotide, utilisée jusqu’à récemment, qui présente une exposition relativement élevée aux rayonnements et des protocoles d’acquisition sur plusieurs jours, est aujourd’hui remplacée par la TEP/TDM au gallium 68 DOTATATE/DOTATOC. L’examen TEP montre une irradiation nettement plus faible (environ 30%) et une résolution nettement améliorée par rapport à la méthode scintigraphique. Dans de nombreux centres NET, la scintigraphie a été complètement remplacée par la TEP/TDM au gallium 68 DOTATATE/DOTATOC (Fig. 1) .
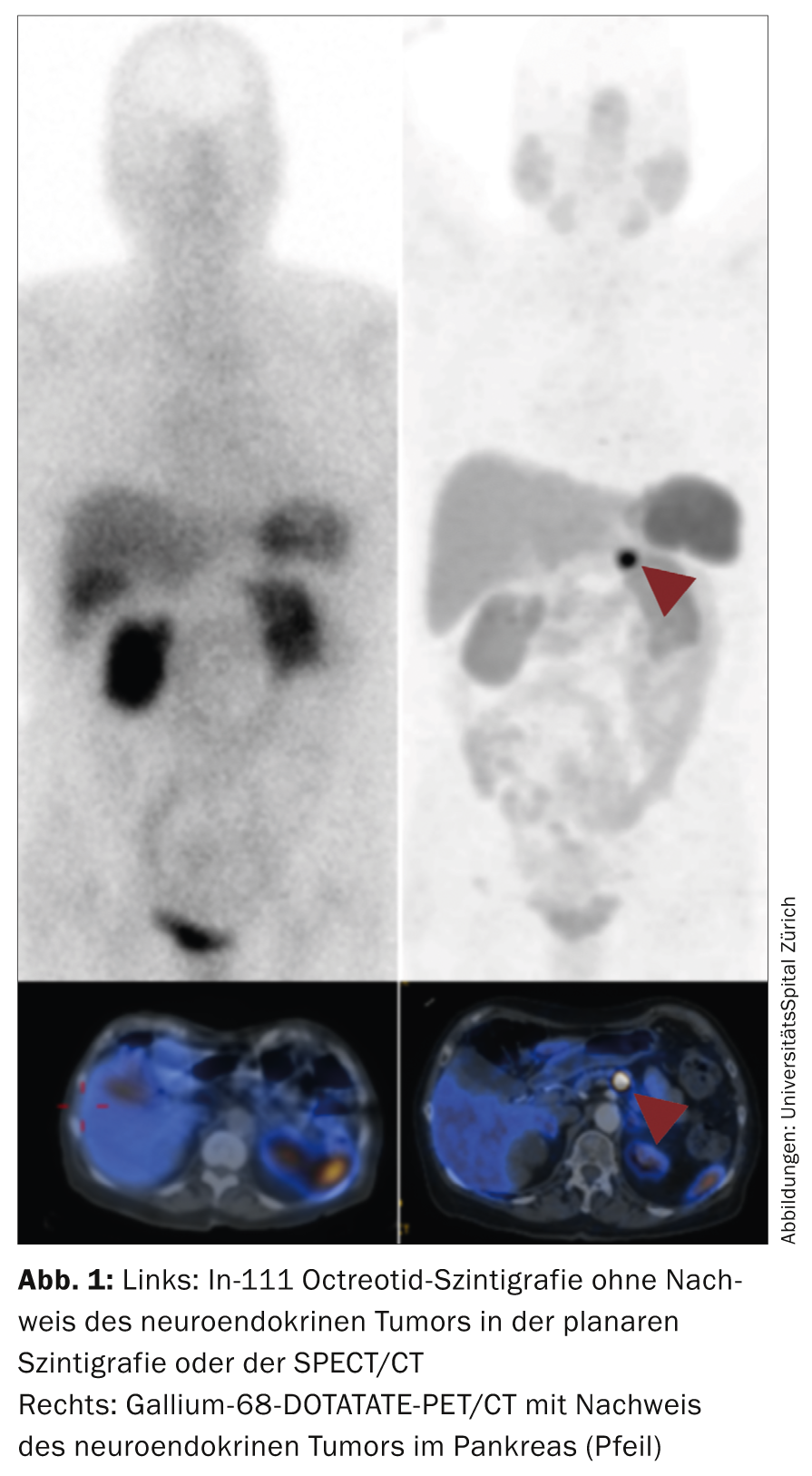
La TEP/TDM au FDG est utilisée pour les cancers dédifférenciés (G3), bien que les tumeurs G2 de plus haut grade puissent déjà être mieux visualisées en TEP au FDG. Les patients atteints de tumeurs fonctionnelles présentant des symptômes carcinoïdes doivent subir au moins une fois une échocardiographie afin d’exclure une implication cardiaque [14].
Marqueurs biochimiques
Les GEP-NET, en particulier les tumeurs “midgut”, peuvent produire et sécréter des peptides fonctionnellement actifs. Cependant, le taux de tumeurs cliniquement fonctionnelles vs. non fonctionnelles n’est que d’environ 1/10. Les tumeurs dites “foregut” et “hindgut” sont un peu moins souvent fonctionnelles. La mesure de ces peptides dans l’urine ou dans le sang peut être utilisée comme marqueur d’évolution en cas de symptômes correspondants [15].
L’examen standard est la mesure sur 24 heures de l’acide 5-indolacétique (5-HIAA) dans l’urine. En présence de symptômes fonctionnels de NET, les taux de sensibilité et de spécificité sont respectivement de à 70 et 90%. La 5-HIAA peut également être augmentée par des maladies inflammatoires chroniques de l’intestin et ainsi influencer le résultat de la mesure de manière faussement positive. De même, les médicaments, par exemple les médicaments de chimiothérapie ou les substances à action centrale comme les antidépresseurs tricycliques, peuvent influencer les valeurs mesurées par le recueil d’urine de 24 heures et doivent être demandés.
D’autres tests, comme le test de jeûne en cas d’insulinome ou le test de sécrétine en cas de gastrinome, doivent être confiés à un spécialiste expérimenté (par exemple, un service d’endocrinologie spécialisé). La chromogranine A (CgA) est un autre marqueur biochimique plasmatique important de la NET. La CgA est produite dans les granules neurosécrétoires des cellules tumorales et peut être sécrétée indifféremment dans les NET fonctionnelles et non fonctionnelles. Il convient de noter que la CgA peut être faussement élevée, en particulier en cas de prise simultanée d’inhibiteurs de la pompe à protons. Les autres facteurs susceptibles d’augmenter la CgA sont : L’insuffisance rénale, la maladie de Parkinson, l’hypertension non traitée, la grossesse, les glucocorticoïdes, la gastrite chronique de type A et les maladies inflammatoires chroniques de l’intestin. Néanmoins, il a été démontré que la CgA est plus précise que la mesure de la 5-HIAA dans l’urine ou que la mesure de l’énolase spécifique des neurones (NSE) fréquemment effectuée [15].
Traitement de la GEP-NET
Le traitement des tumeurs neuroendocrines est complexe et doit être discuté et décidé de manière multidisciplinaire. Un tumor board réunissant différentes disciplines diagnostiques, chirurgicales et médicales devrait décider au moins une fois du diagnostic, du traitement et des contrôles de suivi d’un patient atteint de NET.
La priorité absolue de toute thérapie est l’opération à laquelle on aspire (tant qu’elle semble raisonnable). L’objectif de l’opération doit être, dans la mesure du possible, l’ablation complète de la tumeur. En outre, les tumeurs localement symptomatiques, par exemple en cas de menace d’iléus ou de sang ab ano, doivent être traitées chirurgicalement. Un traitement cytoréducteur peut être discuté dans certains cas dans un cadre palliatif avec une charge tumorale élevée. Toutefois, cela doit être discuté précisément avec le patient compte tenu du contexte palliatif, de la morbidité parfois élevée de l’intervention et de l’évolution souvent lente de la maladie.
Les patients présentant des symptômes fonctionnels prononcés doivent être protégés en périopératoire par de la sandostatine, éventuellement des bloqueurs H1/H2 et de la cortisone [16]. Il convient également de noter que les catécholamines peuvent entraîner une augmentation de la sécrétion d’hormones.
Chez les patients présentant une charge tumorale élevée, les interventions interventionnelles (par exemple l’embolisation) peuvent entraîner une modification rapide de la pression artérielle. Ceci est particulièrement important chez les patients atteints du syndrome carcinoïde avec implication cardiaque. Si une intervention chirurgicale n’est pas possible, on utilise en principe des thérapies locales et interventionnelles ou des thérapies systémiques.
L’embolisation avec des particules, la chimio-embolisation et la radio-embolisation sont utilisées de manière loco-interventionnelle. Cependant, les embolisations à blanc classiques ne sont plus pratiquées qu’en petit nombre dans le cas de la TNE. Ceci est principalement dû au taux élevé d’effets secondaires (en raison de l’effet direct de l’hypoxie locale sévère et de la nécrose). De même, la chimio-embolisation dans la TNE entraîne souvent des symptômes importants (principalement un syndrome post-embolisation), en particulier dans les tumeurs hépatiques avancées et de grande taille. La radioembolisation est une thérapie élégante et généralement sans effets secondaires entre les mains de spécialistes expérimentés. En perfusant localement des microsphères radioactives, typiquement chargées d’yttrium-90, un émetteur β pur, on dépose en une séance des radiations locales allant jusqu’à plus de 500 grays dans les métastases hépatiques. Ceci en raison des propriétés radiatives de l’yttrium-90 et de la vascularisation artérielle sélective typiquement élevée des métastases NET dans le foie (Fig. 2).
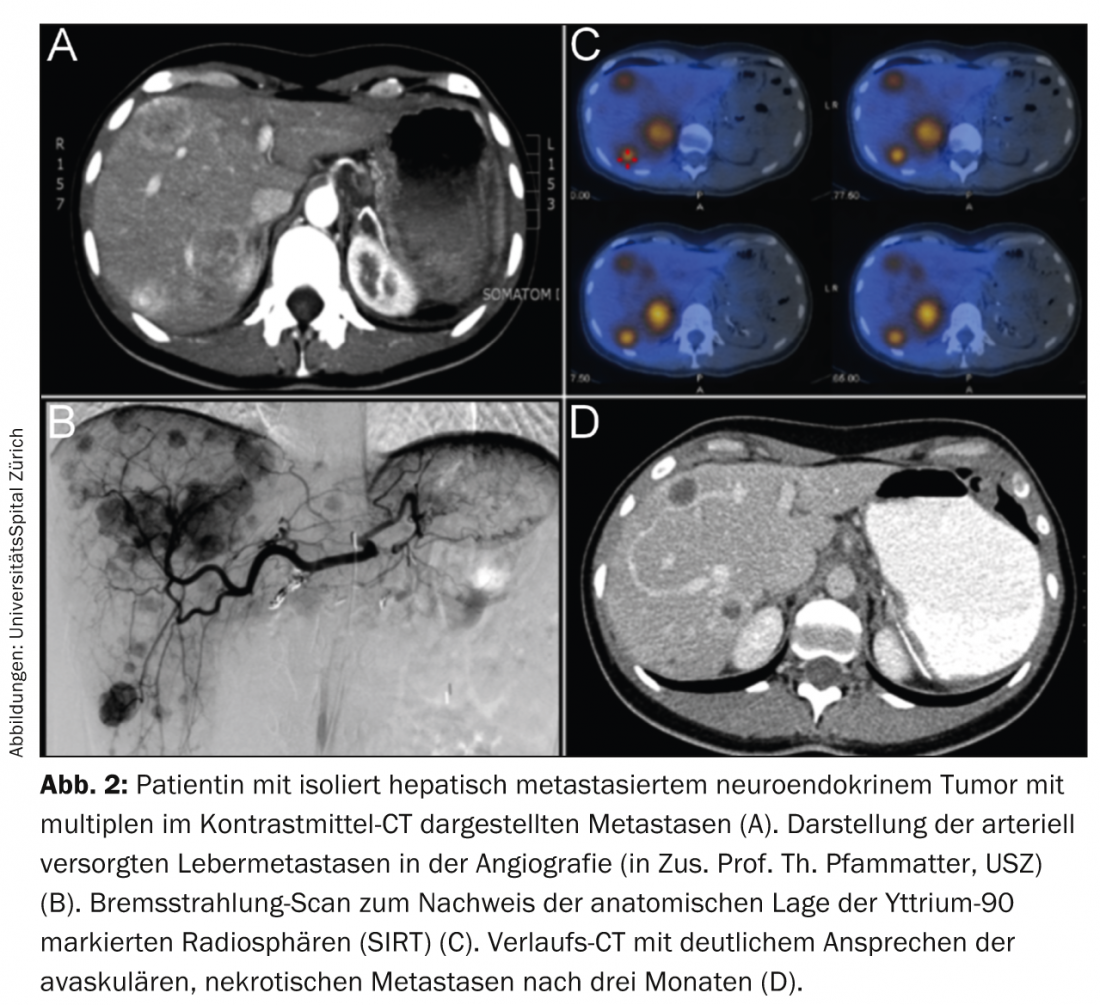
En raison du faible taux d’effets secondaires d’une radioembolisation ambulatoire correctement réalisée, la chimioembolisation et surtout l’embolisation ont été presque entièrement remplacées [17]. Cette thérapie n’est toutefois pratiquée régulièrement que dans quelques centres en Suisse, par exemple le centre d’excellence ENETS de l’hôpital universitaire de Zurich.
Si aucun traitement chirurgical ou systémique local n’est envisageable, un traitement systémique palliatif doit être discuté à la réunion de concertation post-tumorale. Pour les tumeurs non pancréatiques bien différenciées de type “midgut”, le traitement par analogues de la somatostatine est la norme en tant que première ligne de traitement. Il existe en principe deux types d’agents thérapeutiques à longue durée d’action. L’octréotide-LAR a montré une augmentation de la survie sans progression dans les tumeurs “midgut” bien différenciées dans une étude contrôlée par placebo (PROMID-Trial) dans les NET fonctionnellement actives et inactives [18]. L’analyse des données à long terme de l’étude PROMID a également montré une tendance positive de l’octréotide sur l’allongement de la survie globale malgré le cross-over dans le bras de traitement (octréotide) après progression [19].
Récemment, les données du lanréotide ont également été présentées, montrant également une amélioration significative de la survie sans progression [20]. Ces préparations n’ont pas été directement comparées entre elles et il reste à voir, dans la pratique clinique quotidienne, laquelle de ces préparations peut être utilisée dans quelle indication. Tout au plus, le lanréotide sera utilisé dans des tumeurs plus avancées avec une charge tumorale plus élevée, en raison du design de l’étude Clarinet. Dans les tumeurs pancréatiques bien différenciées (peu de tumeurs de “grade intermédiaire”), l’évérolimus a montré une prolongation de la survie sans progression et est autorisé en première ligne dans les p-NET sous le nom d’Afinitor® [21].
L’évérolimus associé à l’octréotide peut également être envisagé dans d’autres TNE, par exemple dans le poumon et le côlon, en raison de son impact positif sur la survie sans progression dans l’étude RADIANT-2, la significativité statistique ayant été manquée de peu [22]. De même, Sutent® montre une augmentation de la survie sans progression et de la survie globale des patients atteints de TNE bien différenciée du pancréas [23]. Ces deux médicaments sont des options pour les patients atteints de p-NET bien différenciés. Cependant, les effets secondaires de ces thérapies sont parfois importants et doivent être effectués par un spécialiste expérimenté. Les thérapies systémiques mentionnées sont principalement utilisées dans les GEP-NET bien différenciés.
Pour les tumeurs G2 plus prolifératives, on utilise classiquement des chimiothérapies combinées. On utilise notamment des thérapies à base de 5-fluorouracile en combinaison avec la streptozotocine ou le Temodal® [24,25]. Ces chimiothérapies sont généralement relativement toxiques, mais peuvent être utilisées avec de bons résultats par des oncologues expérimentés, en particulier chez les patients présentant une charge tumorale élevée, des symptômes et une croissance relativement rapide. Les patients atteints de carcinomes neuroendocriniens à grandes ou petites cellules hautement prolifératifs et dédifférenciés bénéficient souvent d’une chimiothérapie combinée à base de cisplatine et d’étoposide [26]. Les patients présentant une forte expression des récepteurs de la somatostatine à la scintigraphie à l’octréotide ou à la TEP/TDM au gallium 68 DOTATATE/DOTATOC peuvent être évalués pour une thérapie par radionucléides à base de peptides. Cette thérapie par radionucléides administrée par voie systémique irradie les tumeurs SSTR2-positives en fixant et en absorbant des analogues de la somatostatine radiomarqués. Ce traitement fait actuellement l’objet d’un essai de phase III vs. octréotide-LAR. Dans les études de phase II, ce traitement s’est révélé très efficace et avec un très faible taux d’effets secondaires [27]. En particulier, l’insuffisance rénale, souvent mentionnée, survient chez moins de 1% des patients. Cette thérapie est proposée par certains centres en Suisse, par exemple le centre d’excellence ENETS de l’hôpital universitaire de Zurich ou les hôpitaux universitaires de Bâle et de Berne. L’Hôpital universitaire de Bâle a joué un rôle de pionnier dans ce domaine. De nouvelles approches thérapeutiques étudient de nouvelles combinaisons de ces thérapies et explorent de nouvelles voies telles que la réduction des symptômes carcinoïdes [28]. Les analogues de la somatostatine de deuxième génération (Pasireotide Signifor®) n’ont pas montré d’amélioration du contrôle des symptômes dans une étude de phase III, mais une augmentation de cinq mois de la survie sans progression par rapport à l’octréotide LAR [29].
De manière générale, il existe une grande variété de traitements différents pour les GEP-NET et ceux-ci doivent être adaptés à la localisation de la tumeur primaire et à sa différenciation. C’est l’une des raisons pour lesquelles les patients atteints de NET devraient être discutés lors d’un tumorboard spécialisé.
Suivi des patients atteints de NET
Le suivi des patients atteints de TNE varie en fonction de la tumeur primaire et de la différenciation de la tumeur. En général, les insulinomes bénins après ablation, les carcinoïdes rectaux après ablation et les carcinoïdes appendiculaires précoces n’ont pas besoin d’être suivis. Les NET gastriques de type I nécessitent une endoscopie annuelle. En général, les tumeurs G1 nécessitent un contrôle par imagerie et laboratoire tous les douze mois, les tumeurs G2 tous les six mois et les cancers G3 tous les trois mois. Il est recommandé d’utiliser la méthode efficace du staging [30].
Prof. Dr. med. Niklaus G. Schaefer
Littérature :
- Yao JC, et al : One hundred years after “carcinoid” : epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cas in the United States. J Clin Oncol 2008 ; 26(18) : 3063-3072.
- Tsikitis VL, Wertheim BC, Guerrero MA : Trends of Incidence and Survival of Gastrointestinal Neuroendocrine Tumors in the United States : A Seer Analysis. J Cancer 2012 ; 3 : 292-302.
- Rindi G, et al : TNM staging of foregut (neuro)endocrine tumors : a consensus proposal including a grading system. Virchows Archive 2006 ; 4 ; 395-401.
- Rindi G, et al : TNM staging of midgut and hindgut (neuro) endocrine tumors : a consensus proposal including a grading system. Virchows Archive 2007 ; 4 ; 757-762.
- Ruszniewski P, et al : Tumeurs/carcinomes gastriques bien différenciés. Neuroendocrinology 2006 ; 84 : 158-164.
- Lawrence B, et al : A clinical perspective on gastric neuroendocrine neoplasia. Curr Gastroenterol Rep 2011 Feb ; 13(1) : 101-109.
- Robert T, et al. : Tumeur/carcinome duodénal bien différencié (à l’exclusion des gastrinomes). Neuroendocrinology 2006 ; 84 : 165-172.
- Falconi M, et al : Tumeurs/carcinomes pancréatiques non fonctionnels bien différenciés. Neuroendocrinologie 2006 ; 84 : 196-211.
- Eriksson B, et al : Lignes directrices de consensus pour la prise en charge des patients atteints de tumeurs neuroendocrines digestives – Tumeur/carcinome jéjunal iléal bien différencié. Neuroendocrinology 2008 ; 87 : 8-19.
- Ramage JK, et al : Directives de consensus pour la prise en charge des patients atteints de tumeurs neuroendocrines digestives : tumeur/carcinome bien différencié du côlon et du rectum. Neuroendocrinology 2008 ; 87 : 31-39.
- Klöppel G, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Towards a Standardized Approach to the Diagnosis of Gastroenteropancreatic Neuroendocrine Tumors and Their Prognostic Stratification. Neuroendocrinologie 2009 ; 90 : 162-166.
- Sundin A, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Radiological Examinations. Neuroendocrinologie 2009 ; 90 : 167-183.
- Dik J, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Somatostatin Receptor Imaging with 111 In-Pentetreotide. Neuroendocrinology 2009 ; 90 : 184-189.
- Plöckinger U, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Echocardiography. Neuroendocrinology 2009 ; 90 : 190-193.
- O’Toole D, et al. : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Biochemical Markers. Neuroendocrinology 2009 ; 90 : 194-202.
- Akerström G, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Pre- and Perioperative Therapy in Patients with Neuroendocrine Tumors. Neuroendocrinology 2009 ; 90 : 203-208.
- Kennedy A, et al : Radioembolization pour les métastases hépatiques neuroendocrines non résécables en utilisant des microsphères de résine 90Y : résultats précoces chez 148 patients. Am J Clin Oncol 2008 ; 31 : 271-279.
- Rinke A, et al : Étude randomisée, prospective, en double aveugle, contrôlée par placebo, sur l’effet de l’octréotide LAR dans le contrôle de la croissance tumorale chez les patients atteints de tumeurs neuroendocrines métastatiques du mésoblaste : un rapport du groupe d’étude PROMID. J Clin Oncol 2009 Oct 1 ; 27(28) : 4656-4663.
- Arnold R, et al : Étude randomisée, prospective, en double aveugle et contrôlée par placebo sur l’effet de l’octréotide LAR dans le contrôle de la croissance tumorale chez les patients atteints de tumeurs neuroendocrines métastatiques du mésentère (PROMID) : Résultats sur la survie à long terme. J Clin Oncol 2013 ; 31 (suppl ; abstr 4030).
- www.esmo.org/Conferences/Past-Conferences/European-Cancer-Congress-2013/News/Phase-III-Trial-Results-Favour-Lanreotide-Therapy-in-Patients-with-Gastroenteropancreatic-Neuro-Endocrine-Tumours.
- Yao JC, et al : Everolimus pour les tumeurs neuroendocrines pancréatiques avancées. N Engl J Med 2011 Feb 10 ; 364(6) : 514-523.
- Pavel ME, et al : Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2) : a randomised, placebo-controlled, phase 3 study. Lancet 2011 Dec 10 ; 378(9808) : 2005-2012.
- Raymond E, et al : Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011 Feb 10 ; 364(6) : 501-513.
- Engstrom PF, et al : Streptozocin plus fluorouracil versus doxorubicin therapy for metastatic carcinoid tumor. J Clin Oncol 1984 Nov ; 2(11) : 1255-1259.
- Strosberg JR, et al : Chimiothérapie de première ligne à base de capécitabine et de temozolomide chez les patients atteints de carcinomes endocriniens pancréatiques métastatiques. Cancer 2011 Jan 15 ; 117(2) : 268-275.
- Fazio N, Spada F, Giovannini M : Chimiothérapie des cancers neuroendocriniens (NEC) gastro-entéro-pancréatiques (GEP) : un point de vue critique. Cancer Treat Rev 2013 May ; 39(3) : 270-274.
- Kwekkeboom DJ, et al. : Traitement par l’analogue radiolabellisé de la somatostatine [177 Lu-DOTA 0,Tyr3]octreotate : toxicité, efficacité, et survie. J Clin Oncol 2008 mai 1 ; 26(13) : 2124-2130.
- http://telotristat-telestar.com/what-is-the-telestar-trial.php.
- Wolin EM, et al : A multicenter, randomized, blind, phase III study of pasireotide LAR versus octreotide LAR in patients with metastatic neuroendocrine tumors (NET) with disease-related symptoms inadequately controlled by somatostatin analogs. J Clin Oncol 2013 ; 31 (suppl ; abstr 4031).
- Arnold R, et al : ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors : Follow-up and Documentation. Neuroendocrinology 2009 ; 90 : 227-233.
InFo Oncologie & Hématologie 2014 ; 2(5) : 14-18