
Dans le cas du myélome multiple, la clinique menant au diagnostic est dérivée des lésions des organes terminaux. Les critères de diagnostic ont été mis à jour pour la dernière fois en 2014. Avec un diagnostic bien établi et largement disponible, la maladie pose aujourd’hui rarement des problèmes de diagnostic.
Avec une incidence annuelle d’environ 5-6/100 000 et une proportion de 10%, le myélome multiple est l’une des néoplasies hématologiques les plus fréquentes. Avec une médiane d’âge de 65-70 ans, les personnes âgées sont les plus touchées, les hommes étant plus souvent atteints que les femmes avec un ratio de 1,5:1 [1].
Pathogenèse
La transformation néoplasique d’une cellule B du centre germinal en cours de différenciation en plasmocyte producteur d’immunoglobulines est l’événement initiateur de la maladie pour un spectre de maladies des plasmocytes qui se manifestent, selon l’activité de la maladie et la manifestation clinique, comme une gammapathie monoclonale de signification indéterminée (MGUS), un myélome de Smouldering (SM), un myélome multiple (MM) ou une leucémie à plasmocytes (LCP) [2].
La gammapathie monoclonale de signification incertaine est une lésion précurseur clonale dont l’incidence augmente avec l’âge (environ 3% des >70 ans [3]), mais qui n’évolue vers un myélome multiple que dans environ 1% des cas par an [4,5]. Le risque exact de progression dépend du type et de la concentration de la paraprotéine, du ratio des chaînes légères libres, de la proportion de plasmocytes clonaux dans la moelle osseuse et de l’immunoparésie [6,7].
Entre les deux, le stade de myélome asymptomatique dit de Smouldering peut être délimité chez environ 14% des patients, avec un taux de progression annuel de 10% au cours des cinq premières années après le diagnostic initial, suivi de 3% par an au cours des cinq années suivantes et de 1,5% dans les années suivantes [8,9]. Il s’agit d’un état pathologique défini cliniquement entre les MGUS et le myélome multiple, qui comprend un groupe de patients très hétérogène, y compris des patients présentant une évolution prémaligne de type MGUS et des patients atteints de myélome multiple agressif CRAB négatif.
La leucémie à plasmocytes est la forme la plus agressive et leucémique de néoplasie des plasmocytes. Elle est relativement rare, avec une incidence d’environ 4/10 000 000 [10], et peut se développer de manière primaire ou secondaire à partir d’un myélome multiple préexistant (1-4% de tous les patients) [11]. Le diagnostic requiert un taux de plasmocytes de 20% ou une concentration de 2000 plasmocytes/µl de sang sur l’hémogramme microscopique différentiel.
L’événement initiateur de l’oncogenèse des dyscrasies plasmocytaires se déroule dans une phase du développement des cellules B qui, par définition, est marquée par une instabilité génétique due au changement de classe d’isotype de la molécule d’immunoglobuline et à l’hypermutation somatique dans le but d’une maturation par affinité [12].
Sur le plan cytogénétique, on peut distinguer principalement deux modifications du caryotype qui sont présentes, au sens de mutations primaires, très tôt dans l’oncogenèse au stade MGUS. Les caryotypes hyperdiploïdes, observés dans près de deux tiers des cas, sont caractérisés par des trisomies dans les chromosomes de nombre impair (3,5,7,9,11,15,19) et se distinguent du caryotype dit non-hyperdiploïde, qui est souvent dû à des translocations du locus d’immunoglobuline de chaîne lourde (IgH) avec des oncogènes tels que, entre autres, FGFR-3 et MMSET (t[4;14]), MAF (t[14;16]), CCND1 (t[11;14]) ou caractérisé par des gains/pertes non équilibrés de 1q, 1p, 6q, 8p, 13q, 16q et 17p.
Les modifications secondaires comprennent des mutations dans les protéines RAS (K/N-RAS), des mutations activatrices dans des kinases telles que PI3K, AKT, BRAF, des translocations avec activation de facteurs de transcription tels que MYC et des délétions ou inactivation de gènes suppresseurs de tumeurs tels que p53 et RB1 [13].
La maladie se caractérise par une hétérogénéité clonale et une instabilité génomique croissantes [14], qui peuvent être renforcées par une intervention chimiothérapeutique (par ex. alkylants).
Lorsqu’un clone plasmocytaire malin est établi, des lésions cliniques des organes terminaux se développent au fur et à mesure de l’activité de la maladie. Les lésions osseuses ostéolytiques dues à une résorption osseuse accrue sont le résultat d’un métabolisme osseux dérégulé avec une activité accrue des ostéoclastes et une activité supprimée des ostéoblastes, médiée par une augmentation de l’expression de RANKL (“receptor activator of NF kappa B ligand”), une diminution de l’expression de l’ostéoprotégérine [15] et un environnement de cytokines soutenant les ostéoclastes (augmentation de MIP-1 alpha, IL6, IL3, etc.). Une autre conséquence de ce déséquilibre est la libération de calcium de la substance osseuse, avec pour conséquence une hypercalcémie sérique et des modifications de l’excitabilité neuromusculaire.
L’anémie, souvent à l’origine du premier diagnostic, et son exploration sont le résultat du remplacement de l’hématopoïèse saine par des plasmocytes malins. Cependant, cette explication est insuffisante, car on observe souvent des anémies prononcées qui ne peuvent pas être expliquées par une faible infiltration plasmocytaire démontrée. Des modifications du micro-environnement de la moelle osseuse, telles que l’activation de la voie de signalisation du TGFβ, semblent jouer un rôle ici, entraînant une diminution de la concentration de cellules progénitrices hématopoïétiques dans la moelle osseuse des patients atteints de myélome [16].
Outre l’infiltration directe des plasmocytes comme élément pathogène, la paraprotéine monoclonale sécrétée par les plasmocytes joue parfois un rôle déterminant dans la pathogenèse. Le syndrome d’hyperviscosité provoqué par des concentrations élevées de paraprotéines est relativement rare et se retrouve principalement dans les maladies du myélome avec paraprotéine IgM ou IgA, ce qui peut s’expliquer par la structure moléculaire plus complexe de ces dernières, qui se présentent sous forme de pentamères (IgM) ou de dimères (IgA). En revanche, les lésions de l’organe terminal dues à l’amyloïde toxique à chaîne légère dans la pathogenèse de l’amylose AL se produisent même à des concentrations faibles de paraprotéines. Dans le cas du myélome à chaînes légères en particulier, les chaînes légères libres filtrables par les glomérules provoquent des lésions tubulaires et une obstruction (appelée néphropathie de Cast) en raison de la taille réduite de la molécule par rapport à la molécule d’immunoglobuline complète.
Clinique
Les symptômes cliniques qui conduisent finalement à la consultation médicale et à l’établissement du diagnostic découlent des lésions des organes terminaux dues au myélome décrites dans leur pathogenèse. Les douleurs osseuses dues à une ostéolyse liée au myélome, une fracture pathologique d’origine ostéolytique, une diminution des performances en cas d’anémie ou une tendance aux infections conduisent souvent à une consultation médicale. L’effet néphrotoxique des chaînes légères peut entraîner une insuffisance rénale, voire une insuffisance rénale avec une symptomatologie urémique correspondante. Plus rarement, le myélome multiple se manifeste par des troubles du rythme cardiaque, une somnolence ou d’autres symptômes liés à l’hypercalcémie. L’effet toxique de l’amyloïde à chaîne légère dans l’amylose AL peut entraîner une symptomatologie clinique très variée. On y trouve souvent une insuffisance cardiaque due à un dépôt cardiaque, ainsi qu’une insuffisance rénale, une polyneuropathie et autres.
Diagnostic
Les critères de diagnostic du myélome multiple ont été mis à jour pour la dernière fois en 2014 par le Groupe de travail international sur le myélome (IMWG) dans le cadre d’une mise à jour du consensus [17].
Les critères CRAB établis (hypercalcémie, insuffisance rénale, anémie, ostéolyse) ont été complétés par les critères SLiM (tableau 1). L’élargissement des critères diagnostiques est motivé par l’observation que, dans le collectif de patients présentant un myélome de Smouldering ne nécessitant pas de traitement jusqu’à présent, certains paramètres de la maladie sont associés à une forte probabilité de progression (>80% dans les deux ans) en myélome multiple nécessitant un traitement et que ce groupe de patients bénéficie d’une intervention thérapeutique précoce. [17,18].

Ainsi, pour le diagnostic du myélome multiple, la détection de ≥10% de plasmocytes clonaux dans la biopsie ou l’aspiration de moelle osseuse (ill.1) ou un plasmocytome osseux ou extramédullaire documenté par une biopsie, associé à la mise en évidence d’une ou plusieurs lésions d’organes terminaux ou de biomarqueurs de malignité. (Tab.1). La preuve de la clonalité des plasmocytes est apportée par la détection en cytométrie de flux d’une restriction des chaînes légères cytoplasmiques. (ill. 2) ou par coloration immunohistochimique des chaînes légères sur une biopsie représentative de la moelle osseuse.
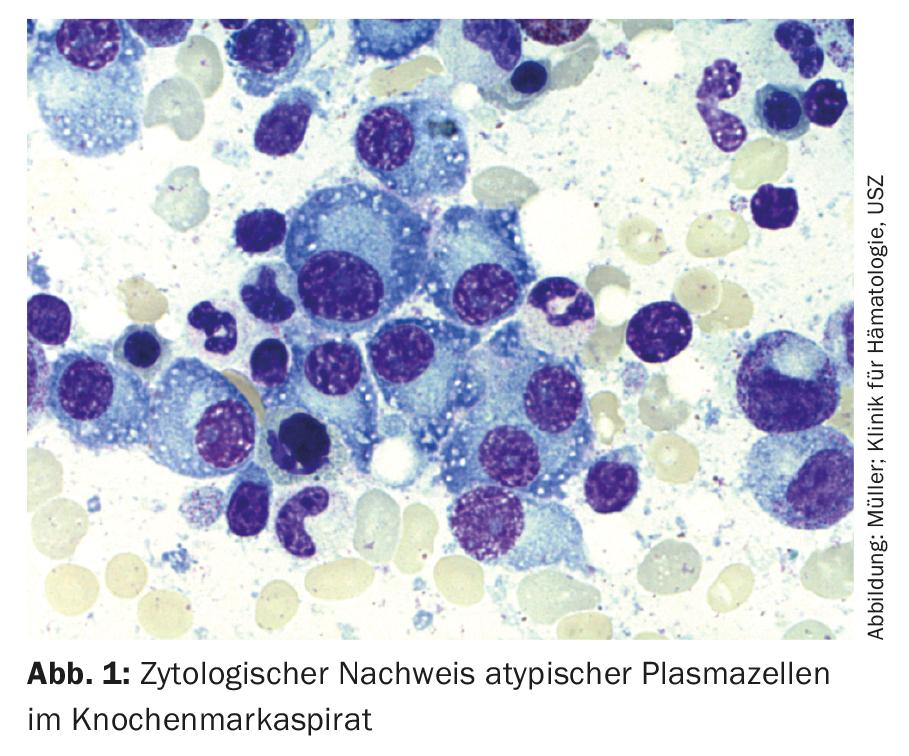
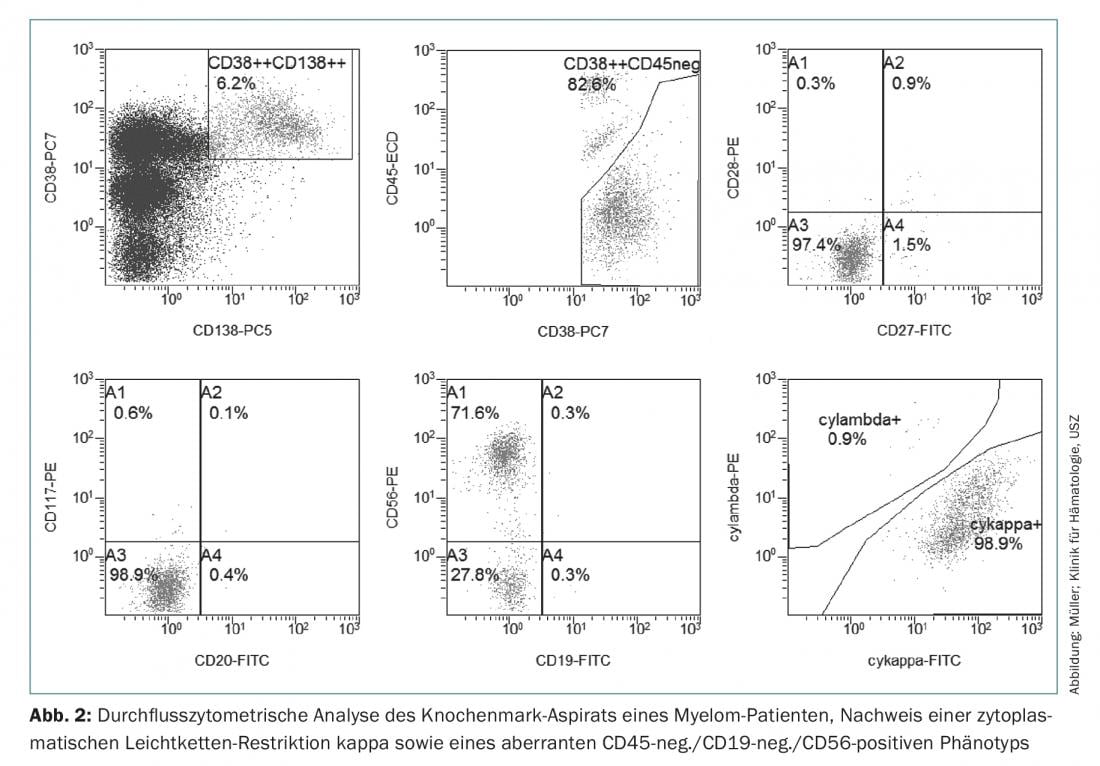
D’un point de vue immunophénotypique, les cellules myélomateuses se distinguent des plasmocytes sains CD38++, CD138+, CD19+, CD45+ et CD56-négatifs par la négativité de CD45 ou une expression réduite, une perte de CD19 et l’expression de marqueurs aberrants tels que CD56, CD117 ou CD28, ce qui peut être exploité dans le diagnostic initial, mais aussi et surtout dans le diagnostic de la MRD, et a parfois une signification pronostique [19].
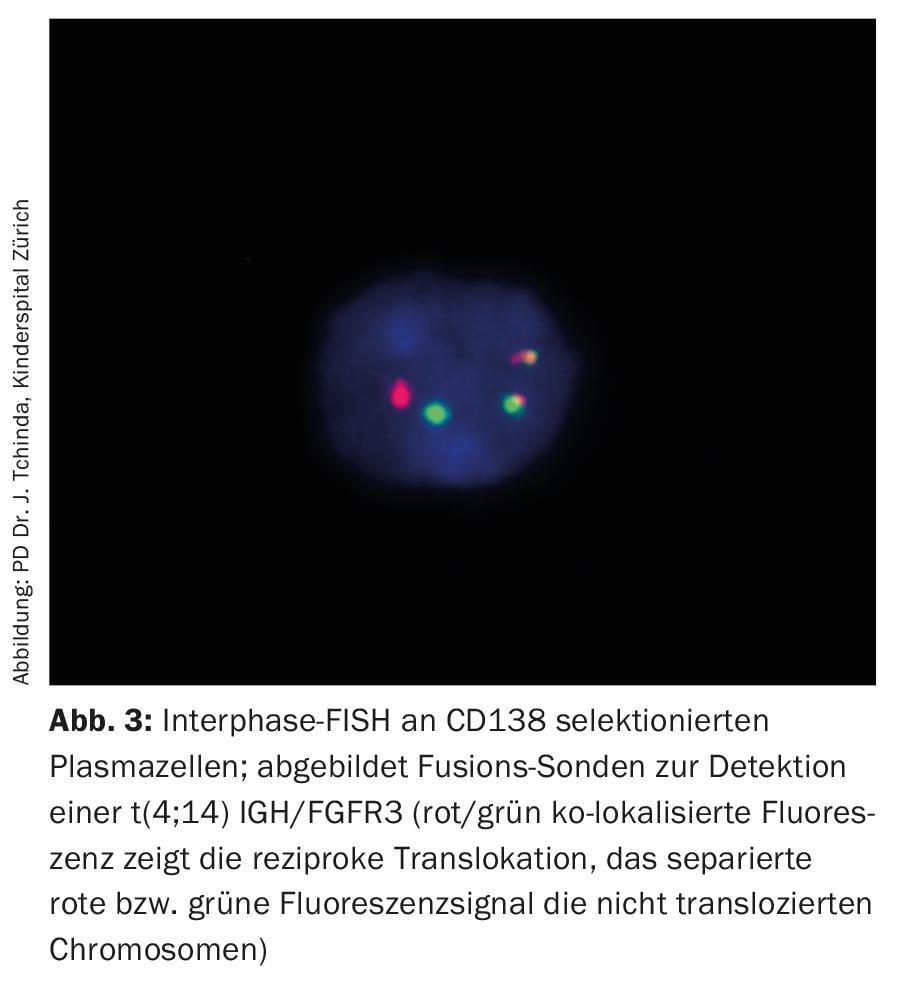
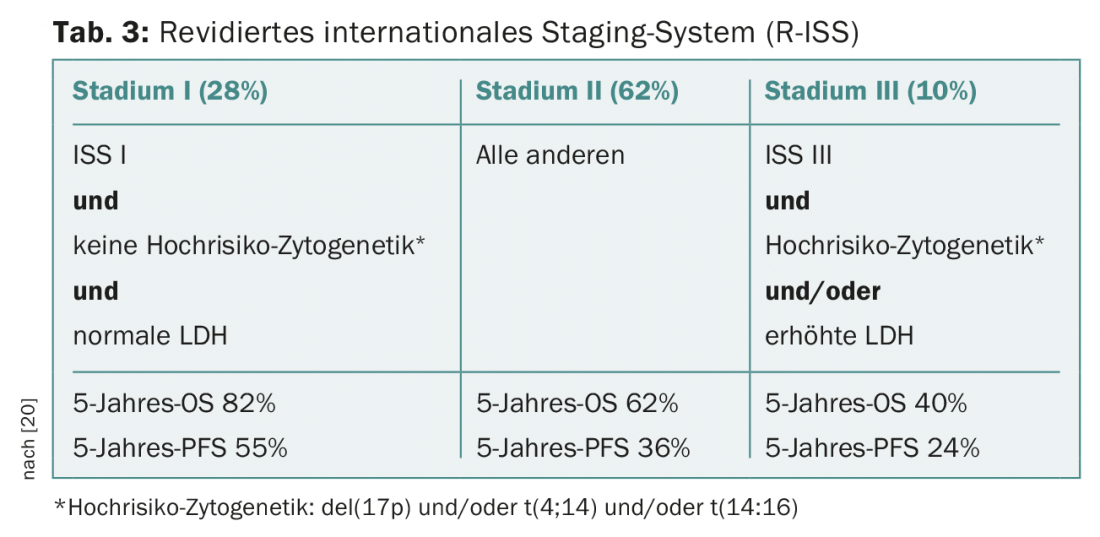
Alors que l’analyse conventionnelle du caryotype des métaphases arrêtées dans le myélome multiple reste généralement peu informative en raison du faible indice de prolifération et des conditions de culture cellulaire difficiles, l’hybridation in situ en fluorescence des noyaux interphasiques avec des sondes fluorescentes fait partie de l’équipement standard et permet, avec les paramètres de substitution établis de l’activité de la maladie comme la β2-microglobuline, l’albumine et la lactate déshydrogénase (LDH), une stratification du risque a priori (Fig. 3, Tab. 3) [20].
Le tableau 2 donne un aperçu des diagnostics de laboratoire à effectuer lors du diagnostic initial ainsi que de l’imagerie nécessaire au moyen d’une TDM à faible dose du corps entier et, en option, d’une IRM et d’une TEP-TDM.
Avec un diagnostic bien établi et largement disponible, le myélome multiple pose rarement des problèmes de diagnostic à l’heure actuelle. Les défis futurs consistent plutôt en une sous-classification génétique de plus en plus précise de l’entité de la maladie, dans le but d’améliorer la stratification du risque et d’établir des marqueurs prédictifs de la réponse au traitement.
Dans le contexte d’options thérapeutiques de plus en plus efficaces avec une meilleure réponse et des rémissions plus profondes, la détection d’une maladie résiduelle minimale (MRD) par cytométrie en flux et séquençage de nouvelle génération joue en outre un rôle de plus en plus important dans l’évaluation de la réponse et de la rémission. Alors que l’importance pronostique de la MRD pour la survie sans progression (PFS) et la survie globale (OS) a déjà été démontrée [21], les décisions thérapeutiques cliniques basées sur la MRD ne sont pas encore établies, mais font l’objet d’études cliniques actuelles.
Messages Take-Home
- La clinique conduisant à la consultation médicale et à l’établissement du diagnostic découle des lésions des organes terminaux liées au myélome.
- Les critères de diagnostic du myélome multiple ont été mis à jour pour la dernière fois en 2014 dans le cadre d’une mise à jour du consensus.
- Avec un diagnostic bien établi et largement disponible, le myélome multiple pose aujourd’hui rarement des problèmes de diagnostic.
- Le défi futur réside plutôt dans une sous-classification génétique de plus en plus précise de l’entité de la maladie, dans le but d’améliorer la stratification du risque et d’établir des marqueurs prédictifs de la réponse au traitement.
- L’importance pronostique de la maladie résiduelle minimale (MRD) pour la survie sans progression (PFS) et la survie globale (OS) a été démontrée, les décisions thérapeutiques cliniques basées sur la MRD font l’objet d’études cliniques actuelles.
Littérature :
- Rodriguez-Abreu D, Bordoni A, Zucca E : Épidémiologie des tumeurs hématologiques. Annals of Oncology 2007 ; 18(Suppl 1) : i3-i8.
- Bakkus MH, et al : Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood 1992 ; 80 : 2326-2335.
- Kyle RA, et al : Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006 ; 354 : 1362-1369.
- Landgren O, et al : La gammopathie monoclonale de signification indéterminée (MGUS) précède généralement les myélomes multiples : une étude prospective. Blood 2009 ; 113 : 5412-5417.
- Weiss BM, et al : Une gammapathie monoclonale précède un myélome multiple chez la plupart des patients. Blood 2009 ; 113 : 5418-5422.
- Baldini L, et al : Role of different hematologic variables in defining the risk of malignant transformation in monoclonal gammopathy. Blood 1996 ; 87 : 912-918.
- Turesson I, et al. : Gammopathie monoclonale de signification indéterminée et risque de malignité lymphoïde et myéloïde : 728 cas suivis jusqu’à 30 ans en Suède. Blood 2014 ; 123 : 338-345.
- Kristinsson SY, Holmberg E, Blimark C : Traitement du myélome smoldering à haut risque. N Engl J Med 2013 ; 369 : 1762-1763.
- Kyle RA, et al : Clinical course and prognostic of smoldering (asymptomatic) multiple myeloma. N Engl J Med 2007 ; 356 : 2582-2590.
- Sant M, et al : Incidence des tumeurs hématologiques en Europe par sous-type morphologique : résultats du projet HAEMACARE. Blood 2010 ; 116 : 3724-3734.
- Tiedemann RE, et al : Aberrations génétiques et survie dans la leucémie à cellules plasmatiques. Leucémie 2008 ; 22 : 1044-1052.
- Seifert M, Scholtysik R, Küppers R : Origine et pathogenèse des lymphomes à cellules B. Méthodes Mol Biol 2013 ; 971 : 1-25.
- Kuehl WM, Bergsagel PL : Myélome multiple : évolution des événements génétiques et des interactions avec l’hôte. Nat Rev Cancer 2002 ; 2 : 175-187.
- Bolli N, et al : Hétérogénéité de l’évolution génomique et des profils mutationnels dans le myélome multiple. Nat Commun 2014 ; 5 : 2997.
- Roodman GD : Mécanismes des métastases osseuses. N Engl J Med 2004 ; 350 : 1655-1664.
- Bruns I, et al : Multiple myeloma-related deregulation of bone marrow-derived CD34(+) hematopoietic stem and progenitor cells. Blood 2012 ; 120 : 2620-2630.
- Rajkumar SV, et al : International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014 ; 15 : e538-48.
- Mateos MV, et al : Lenalidomide plus Dexamethasone pour le myélome multiple à haut risque. N Engl J Med 2013 ; 369 : 438-447.
- Mateo G, et al : Valeur pronostique de l’immunophénotypage dans le myélome multiple : une étude par les groupes d’étude coopératifs PETHEMA/GEM sur les patients traités de manière uniforme avec un traitement à haute dose. Journal of Clinical Oncology 2008 ; 26 : 2737-2744.
- Palumbo A, et al : Révision du système international de stadification du myélome multiple : un rapport du groupe de travail international sur le myélome. J Clin Oncol 2015 Sep 10 ; 33(26) : 2863-2869.
- Paiva B, van Dongen JJM, Orfao A : Nouveaux critères d’évaluation de la réponse : rôle de la maladie résiduelle minimale dans le myélome multiple. Blood 2015 ; 125 : 3059-3068.
- Moreau P, et al : Myélome multiple : ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up ? Ann Oncol 2017 Jul 1 ; 28(suppl_4) : iv52-iv61.
InFo ONKOLOGIE & HÉMATOLOGIE 2017 ; 5(5) : 7-10









