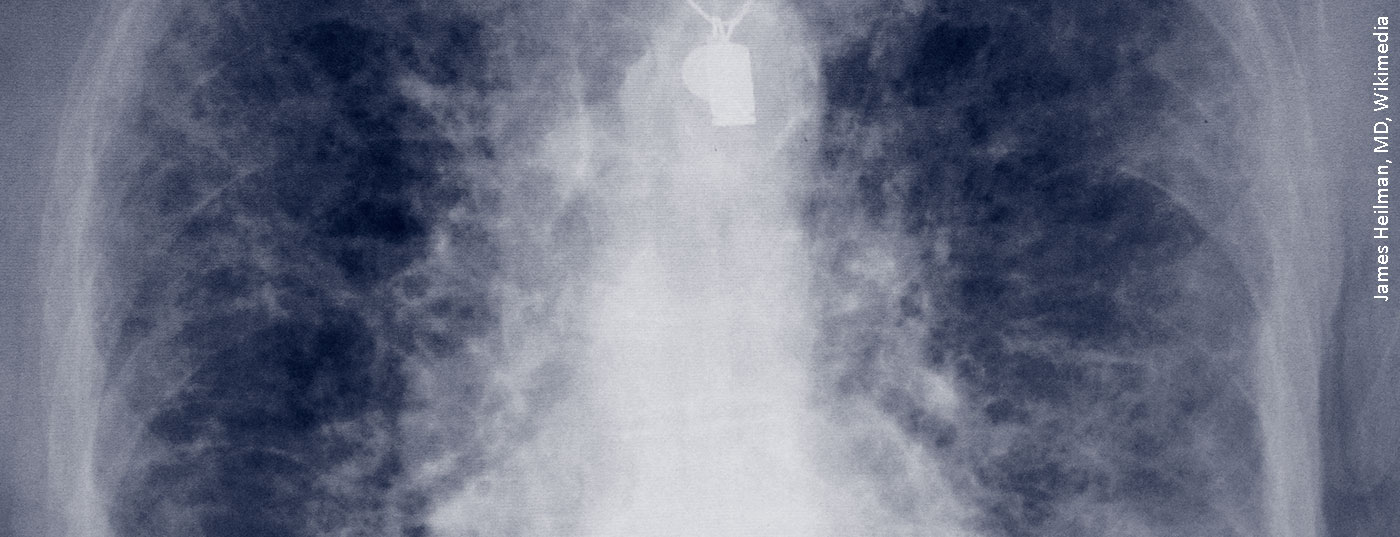
La sclérose systémique (SSc) est l’une des maladies les plus graves, car elle se manifeste partout dans le corps, mais il n’y a pas de solution à peu de problèmes et pratiquement chaque organe doit être traité séparément. Il n’existe toujours pas de traitement universel comme pour d’autres maladies, c’est-à-dire un médicament biologique ou un immunosuppresseur spécifique.
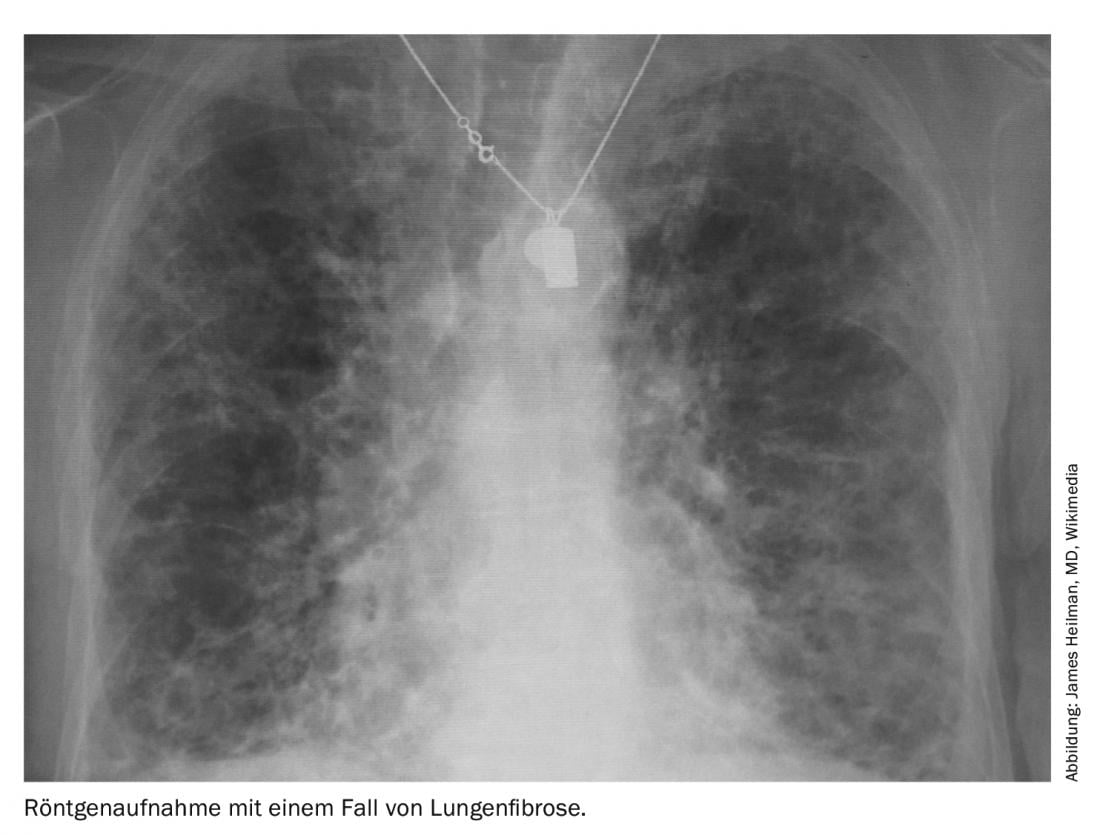
L’examen des poumons est obligatoire pour tout patient se présentant avec une sclérose systémique, et ce pour deux composantes : d’une part la fibrose pulmonaire, qui est pire, et d’autre part l’hypertension pulmonaire (HTAP), pour laquelle il existe au moins plus de médicaments. La perte de la capacité d’expansion des poumons et la perte de leur fonction d’échange gazeux sont les deux mécanismes pathologiques de la fibrose pulmonaire. Elle est 100 fois moins fréquente, mais fait plus de morts que l’asthme chaque année. Le dilemme réside dans le fait qu’il existe plus d’une centaine de pathologies différentes qui peuvent aboutir au stade final de la fibrose pulmonaire.
“L’aggravation peut être très rapide dans la sclérose systémique, et c’est ce qui est dangereux”, a averti le professeur Ulf Müller-Ladner, du département de rhumatologie et d’immunologie clinique de la clinique Kerckhoff de Bad Nauheim (Allemagne). “Les patients n’ont pas de grands signes avant-coureurs, peut-être un ulcère digital, et soudain la fibrose pulmonaire ou l’hypertension pulmonaire est déjà là”. Les patients eux-mêmes ne s’en aperçoivent que lorsque des troubles dyspnéiques apparaissent. C’est pourquoi, selon le rhumatologue, il est également élémentaire dans sa discipline de toujours examiner les poumons d’un patient, qu’il présente ou non des symptômes.
Dans la SSc, la fibrose pulmonaire a pris une place de plus en plus importante dans la liste des manifestations organiques. En effet, d’autres complications telles que les crises rénales ou les problèmes cardiovasculaires ou gastro-intestinaux sont devenues plus faciles à traiter avec le temps, contrairement à la fibrose pulmonaire associée à la SSc (SSc-ILD). Le professeur Andreas Günther, directeur de l’unité de recherche sur les maladies pulmonaires fibrosantes à l’hôpital universitaire de Giessen (Marburg), a déclaré : “Si l’on stratifie sur la base des dimensions en HRCT, plus de 30% des patients atteints de SSc présentent une fibrose au scanner”. “Il s’agit donc déjà plutôt d’une maladie très prononcée avec un mauvais résultat”.
Meilleur suivi = résultats mieux contrôlés
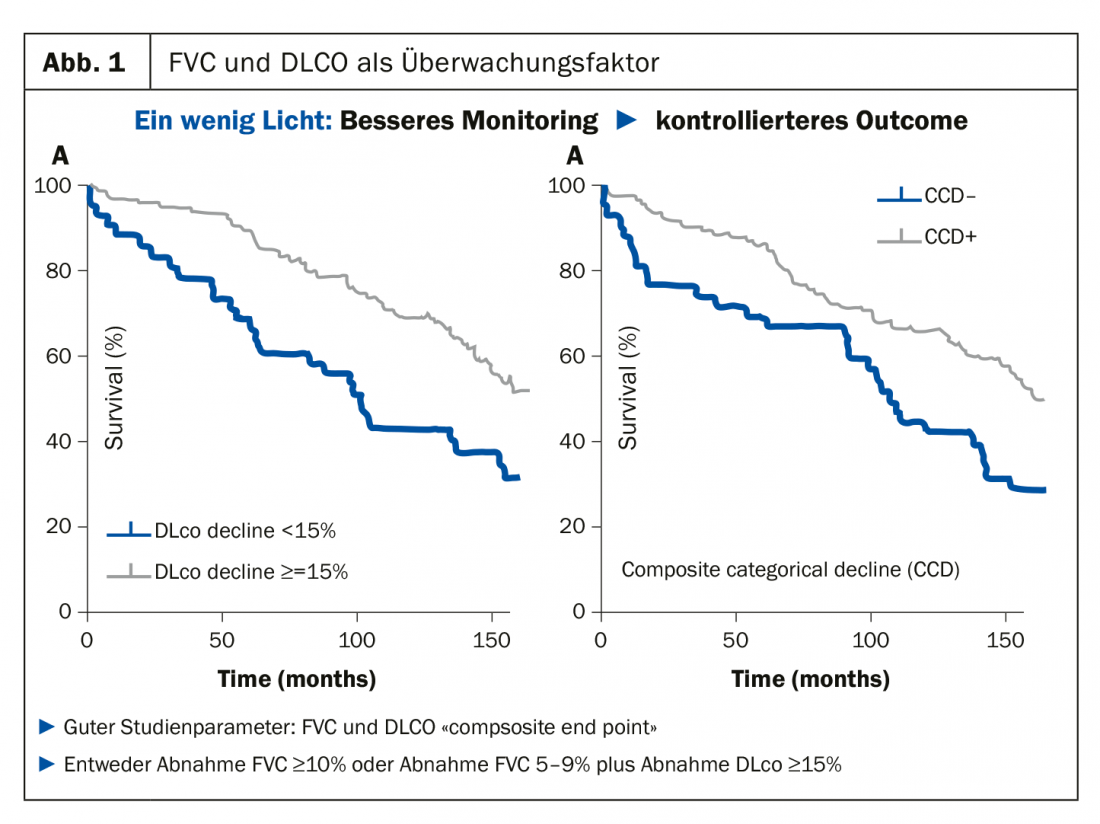
En tant que rhumatologue qui doit surveiller les poumons, le professeur Müller-Ladner conseille toujours à ses collègues de s’en tenir aux paramètres relativement simples à mesurer, c’est-à-dire la capacité vitale (CVF) et la capacité de diffusion (DLCO), car c’est justement cette combinaison qui s’est développée de manière très positive dans les études en tant que facteur de surveillance. “Si la combinaison de ces deux paramètres progresse très rapidement, les courbes s’écartent également” (Fig. 1). Ce point final composite est désormais bien établi dans les études. La diminution de la FVC ≥10% ou une diminution de 5-9% plus une diminution de la DLCO de ≥15% doivent être considérées comme déterminantes. “En tant que rhumatologues, ces petits chiffres peuvent nous faire sourire, mais en ce qui concerne les poumons, ils ont une valeur absolue”, a expliqué le spécialiste en mettant en évidence un facteur que ses collègues spécialistes ont tendance à sous-estimer : “Un petit changement au niveau des poumons peut avoir de grandes conséquences, et les patients le remarquent”.
“L’important, c’est de faire quelque chose”
Dans de nombreuses maladies rhumatologiques, le traitement est similaire : il y a des cellules inflammatoires que l’on met au repos, et tout va bien – sans compter les effets secondaires. Pour la SSc, la situation est différente. “Parce que vous avez ici plusieurs composants qui fonctionnent en parallèle et que personne ne sait vraiment par quoi cela commence – le système immunitaire, la fibrose, les vaisseaux ?” Et si vous baissez l’un de ces interrupteurs, vous pouvez éventuellement augmenter quelque chose à un autre endroit, ce qui n’améliore pas l’état général. En même temps, vous avez affaire à une fibrose, “et antagoniser une fibrose est l’une des tâches les plus difficiles qui soient”. La question reste donc de savoir comment procéder dans le traitement : supprimer le déclencheur ou établir un lien direct ?
Les rhumatologues répondent à cette question en faisant ce qu’ils peuvent et savent faire, selon le professeur Müller-Ladner : ils prennent du méthotrexate ou de la pénicillamine, “l’essentiel est de faire quelque chose”. Mais parfois, l’activisme peut aussi faire des dégâts, par exemple si le diagnostic n’est pas bon. En cas de doute, il est donc conseillé de consulter un pneumologue. Le professeur Günther a fait référence aux directives de traitement EULAR/EUSTAR SSc-ILD de 2016, qui recommandent le cyclophosphamide ou une autogreffe de cellules souches hématopoïétiques (HSCT) (tab. 1). Le mycophénolate mofétil (MMF), selon le commentaire du pneumologue, n’est certainement pas moins efficace que le cyclophosphamide, mais il est moins toxique, raison pour laquelle il est encore volontiers prescrit.

Des études prometteuses
Autre problème : la sclérose systémique est et reste une maladie rare, ce qui rend les grandes études souhaitables mais rares. Le professeur Müller-Ladner a expliqué : “Il n’est pas possible de secouer le poignet comme pour la polyarthrite rhumatoïde (PR), parce que vous n’arrivez tout simplement pas à obtenir les chiffres”. C’est pourquoi chaque publication est précieuse. Le rhumatologue a d’abord présenté deux études (FaSScinate et FocuSSced) avec un nombre relativement faible de participants, qui mettaient toutes deux l’accent sur la lutte contre l’inflammation avec l’interleukine-6 (IL-6) au moyen du tocilizumab. L’étude FocuSSced, en particulier, a montré qu’à long terme, l’inhibition de l’inflammation par rapport à la CVF sous IL-6 était prometteuse.
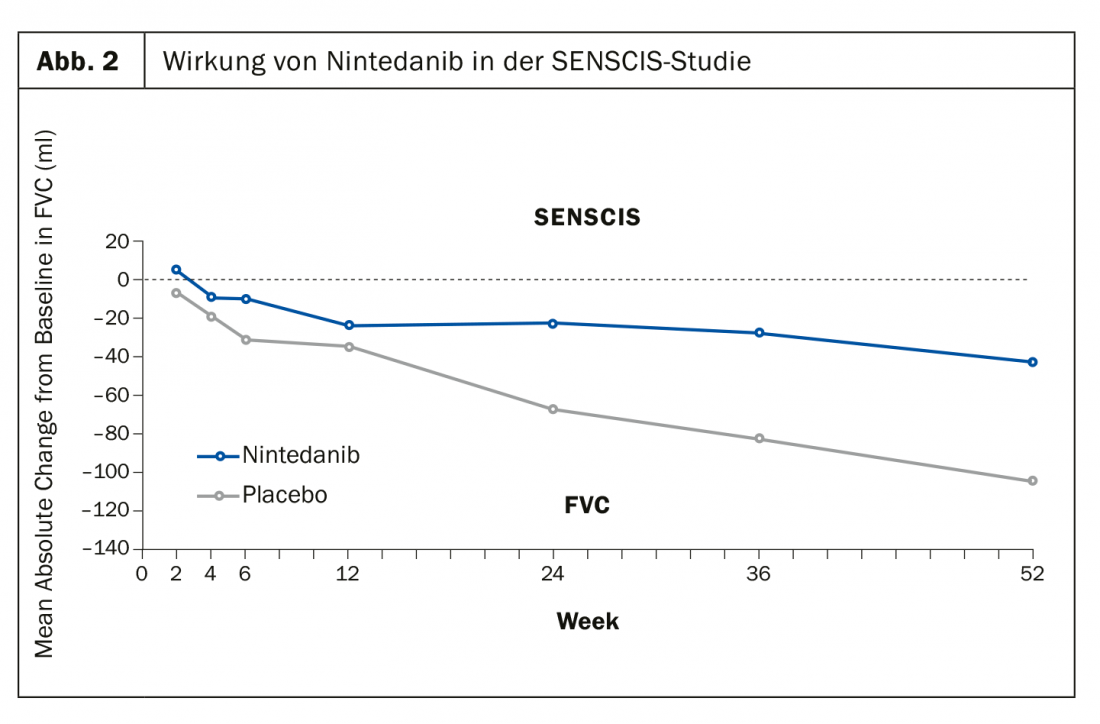
L’étude SENSCIS, quant à elle, a évalué l’effet du nintedanib par rapport au placebo avec comme critère d’évaluation principal la réduction de la capacité vitale fonctionnelle (mL/an). Elle est axée sur une médication de base stabilisée en vie réelle avec du MTX ou du MMF. Là encore, les résultats étaient positifs après 52 semaines (fig. 2). “Nous avons besoin d’un antifibrotique raisonnable”, a déclaré le professeur Müller-Ladner avec confiance, “car jusqu’à présent, la fibrose ne peut être traitée que de manière indirecte”.
Cependant, le nintédanib n’est actuellement pas approuvé pour le traitement de la SSc/SSc-ILD ou d’autres pneumopathies interstitielles progressives (PF-ILD) en dehors de la fibrose pulmonaire idiopathique (FPI). Il est d’autant plus important, selon lui, d’obtenir dans un avenir proche une autorisation de mise sur le marché qui ne se limite pas aux formes purement pulmonaires de la maladie.
Source : Symposium industriel “Fibrose pulmonaire : un défi pour la SSc et d’autres maladies rhumatismales” dans le cadre du DGRh ; Organisateur : Boehringer Ingelheim
InFo PNEUMOLOGIE & ALLERGOLOGIE 2019 ; 3(1) : 30-31 (publié le 11.12.19, ahead of print)