La néoplasie myéloproliférative des cellules souches hématopoïétiques est principalement causée par des mutations somatiques du gène JAK2 et l’hématopoïèse autonome clonale qui en résulte. Le risque de myélofibrose ou de transformations leucémiques a une incidence sur le pronostic.
La polycythemia vera (PV) est actuellement classée par l’Organisation mondiale de la santé (OMS) dans la catégorie principale des néoplasmes myéloprolifératifs (NMP) [1,2]. Dans l’état actuel des connaissances, la PV est une maladie des cellules souches ou progénitrices hématopoïétiques, principalement due à des mutations somatiques du gène JAK2, avec une myéloprolifération clonale qui en résulte [3]. De rares cas familiaux ont été décrits, ceux-ci présentant des mutations germinales dans le récepteur de l’érythropoïétine (EPOR) [4]. L’incidence de la PV en Europe est de 0,4 à 2,8% d’habitants par an. L’âge moyen de la maladie se situe entre 60 et 65 ans, les femmes et les hommes sont touchés à peu près à égalité [5,6]. La durée de survie moyenne est de 14 ans chez les patients PV âgés (>60 ans) et de 24 ans chez les moins de 60 ans [7].
Clinique
Dans l’hémogramme, la maladie se manifeste par une augmentation de la production d’érythrocytes (érythrocytose) indépendamment des mécanismes de régulation physiologiques. Une leucocytose et une thrombocytose dues à une augmentation de la mégacaryopoïèse sont souvent associées. Au cours de l’évolution de la maladie, jusqu’à 10% des patients PV passent par une phase dite de “retard”, caractérisée par une hématopoïèse réduite, en raison de la progression des altérations fibrotiques, au sens d’une myélofibrose [8]. Une transformation leucémique est diagnostiquée dans jusqu’à 5% des cas de PV après 20 ans d’évolution de la maladie [7,9]. Les facteurs de risque pronostiques de myélofibrose, de transformation leucémique et de réduction de la survie globale sont un âge élevé du patient, une leucocytose, une thrombocytose et un caryotype anormal [10].
La survenue d’événements thromboemboliques veineux et artériels chez 20 à 50% des patients atteints de PV est également d’une importance pronostique décisive. D’un point de vue physiopathologique, elles sont dues à une augmentation de la viscosité sanguine et aux modifications rhéologiques qui en résultent, ainsi qu’à des stimuli inflammatoires, procoagulants et microvasculaires [11,12]. On distingue deux groupes de risque de thromboses récurrentes : Un groupe à haut risque chez les patients >60 ans et ayant des antécédents positifs d’événements thrombotiques et un groupe à bas risque en l’absence des deux facteurs [13]. L’hypertension artérielle est un facteur de risque indépendant pour l’apparition de thrombus artériels.
Cliniquement, la PV se manifeste principalement par de la fatigue et de l’épuisement (jusqu’à 84,9%) [14] et du prurit (dans environ 40%) [15]. Les autres symptômes cliniques comprennent une rougeur du visage, une peau et des muqueuses bleu-rouge, une pression sur la tête, des maux de tête et une hypertension. Les troubles microcirculatoires entraînent souvent des symptômes cliniques caractéristiques (par ex. troubles visuels, paresthésies, érythromélalgie, troubles pectangineux) [16]. Les thromboses veineuses abdominales et les thromboses veineuses sinusales sont d’autres complications aux conséquences graves et souvent mortelles [17,18]. Paradoxalement, dans de rares cas de thrombocytose extrême (>1000×109/L), on observe également une tendance accrue aux saignements. La cause en est une diminution du facteur Von Willebrand (VWF) dans le sens d’un syndrome de consommation acquis. Sur le plan pathobiologique, une activité ADAMTS13 accrue ainsi qu’une sensibilité plaquettaire accrue et une activation plaquettaire inadéquate qui en résulte jouent un rôle essentiel [19,20]. Une splénomégalie est observée chez environ un tiers des personnes atteintes et peut s’accompagner de douleurs et d’infarctus.
Pathobiologie de la PV
La PV se caractérise par une myéloprolifération clonale due à des altérations pathologiques des cellules souches hématologiques [3]. Chez presque tous les patients atteints de PV, on trouve une mutation de la Janus kinase 2 (JAK2), située sur le locus 9p24, 96% de tous les cas présentant une mutation somatique activatrice dans l’exon 14 (JAK2V617F) [21] et plus rarement (environ 3%) dans l’exon 12 [22]. Jusqu’à présent, aucune différence significative dans l’évolution clinique n’a été démontrée en fonction de la mutation en question [23].
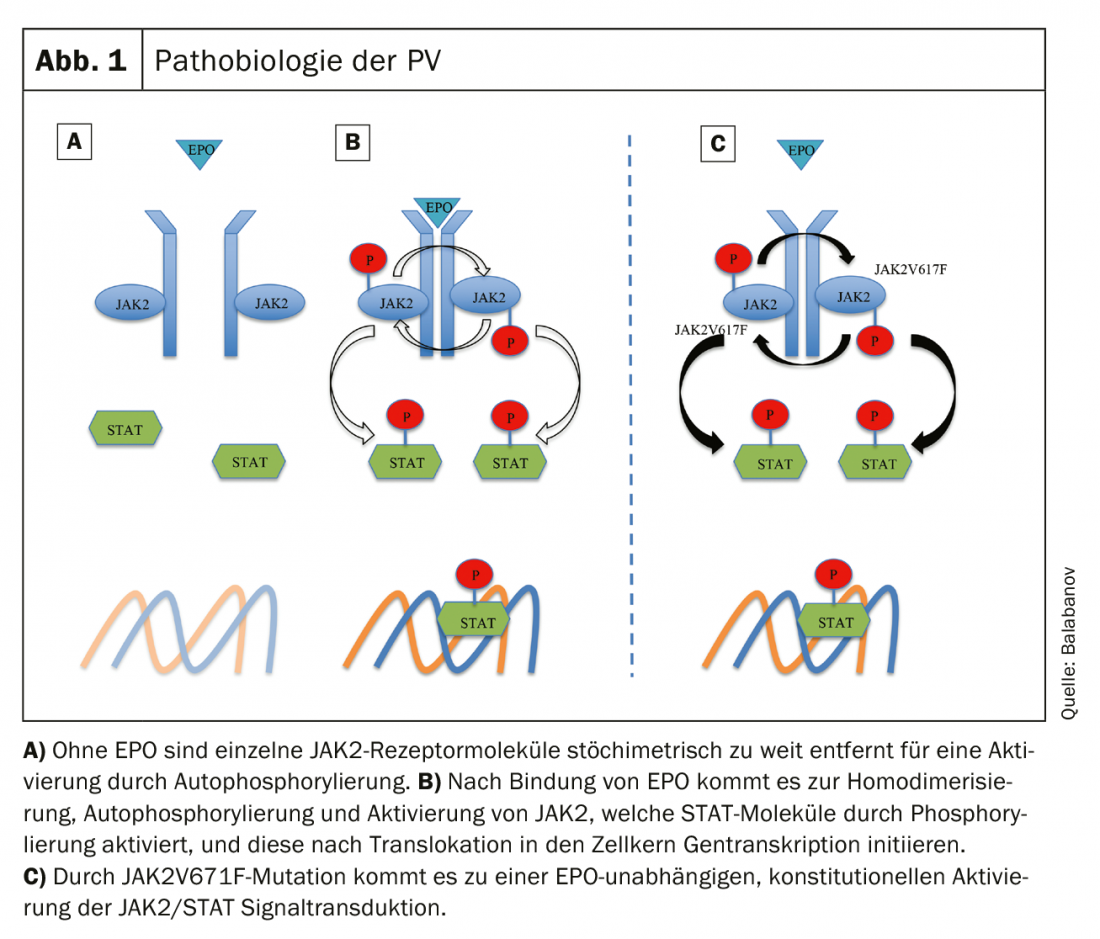
Dans des conditions physiologiques, la liaison de l’érythropoïétine (EPO) au récepteur de l’EPO (EPOR) entraîne un changement de conformation (homodimérisation du récepteur), avec l’autophosphorylation et l’activation de JAK qui en résultent. Les molécules JAK activées phosphorylent et activent ensuite les molécules STAT (signal transducer and activator of transcription) qui, en tant que facteurs de transcription, déclenchent des signaux de croissance hématopoïétique après translocation dans le noyau cellulaire [24]. La mutation activatrice JAK2V617F entraîne une activation constitutionnelle JAK/STAT indépendante de l’EPO et une prolifération cellulaire incontrôlée (figure 1) [25]. D’autres altérations génétiques ont été décrites, comme la dérégulation de l’inhibiteur d’apotose Bcl-x ou du facteur de transcription NF-E2, mais semblent être des conséquences secondaires d’une mutation de JAK2 [26].
Les mutations JAK2V617F sont plus fréquentes chez les patients âgés et sont associées à une panmyélocytose, à une myélofibrose et à des symptômes cliniques tels que des démangeaisons [22,23]. Aucune association avec la survie globale ou le risque de transformation n’a été démontrée [3]. La fréquence des allèles mutés ne semble pas non plus influencer la survie ou la fréquence des transformations leucémiques. Dans une étude de la Mayo Clinic, le ciblage par séquençage profond a permis de montrer que plus de la moitié (53%) des patients PV étudiés présentaient des mutations supplémentaires en plus de JAK2 [28]. Les mutations les plus fréquentes ont été détectées dans TET2 (22%), ASXL1 (12%) et SH2B3 (9%) (d’autres mutations ont été détectées dans SRSF2, IDH2, TP53). Les mutations dans ASXL1, SRSF2 et IHD2, en particulier, ont montré un effet pronostique indépendant des autres prédicteurs (survie médiane de 7,7 ans avec vs. 16,9 ans sans mutations). Un caryotype anormal est présent chez 15% des patients au moment du diagnostic initial, les altérations cytogéniques les plus fréquemment détectées étant -Y, +8, +9, del(20q) et 1q+ [27].
Diagnostic
Une anamnèse détaillée et un examen physique constituent la base du diagnostic. Le questionnaire MPN-SAF (Myeloproliferative Neoplasm Symptom Assessment Form) permet une évaluation structurée et complète des symptômes pertinents [29]. Il convient en particulier de tenir compte des antécédents d’événements thrombotiques, des stigmates de thrombose, de l’hypertension et de l’âge du patient, en raison de l’importance du pronostic et du traitement. Pour évaluer une splénomégalie, il est recommandé de procéder à une échographie abdominale.
En laboratoire, il convient de déterminer la concentration d’EPO en plus d’un hémogramme différentiel et d’un bilan de coagulation. Le diagnostic définitif ne peut être établi qu’à partir d’une biopsie de la moelle osseuse.
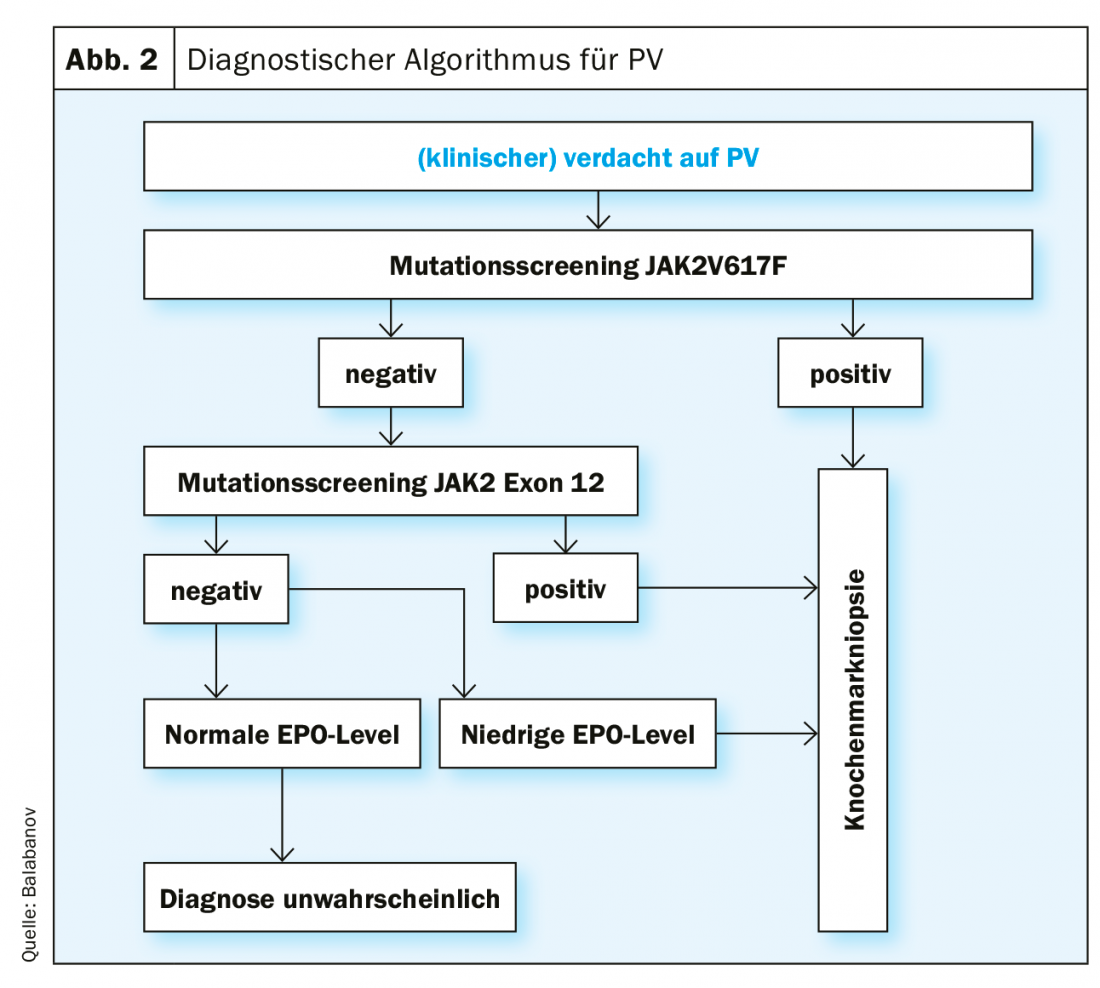
Les critères de diagnostic selon la classification actuelle (2016) de l’OMS sont présentés dans le tableau 1. Pour pouvoir poser un diagnostic de PV, tous les critères principaux ou deux critères principaux et le critère secondaire doivent être remplis. La détection d’une mutation JAK2V617F est extrêmement fiable, avec une sensibilité de test de 97% et une spécificité de près de 100%. En cas de statut de mutation JAK2V617F négatif, mais de niveau d’EPO sérique abaissé, une analyse de mutation dans l’exon 12 du gène JAK2 est également indiquée. En raison de l’importance pronostique pour la survie globale et la survie sans leucémie, une détermination du caryotype devrait être effectuée le cas échéant. En outre, une analyse des mutations par séquençage de nouvelle génération (NGS) est utile, car les mutations dans ASXL1, SRSF2 et IDH2 ont une pertinence pronostique. Cependant, le NGS n’est pas encore un examen standard pour tous les patients atteints de PV au moment du diagnostic. Cependant, une étude pionnière récemment publiée a montré que l’intégration complète de variables génomiques et cliniques permettait une stratification personnalisée du risque avec des implications thérapeutiques [30] (https://cancer.sanger.ac.uk/mpn-multistage).

Un algorithme basé sur les critères diagnostiques actuels de l’OMS pour la procédure diagnostique en cas de suspicion de PV est présenté dans la figure 2. Des tests fonctionnels spécifiques du VWF sont indiqués pour clarifier une tendance accrue aux saignements (par exemple, l’activité du cofacteur ristozitine).
Thérapie
Avec un taux de survie à 10 ans de plus de 75%, ainsi qu’un risque relativement faible de myélofibrose (<10%) et de dégénérescence maligne dans le sens d’une transformation leucémique (<5%) [8], l’objectif principal du traitement de la PV est de prévenir les complications thrombo-hémorragiques et de soulager les symptômes décrits ci-dessus.
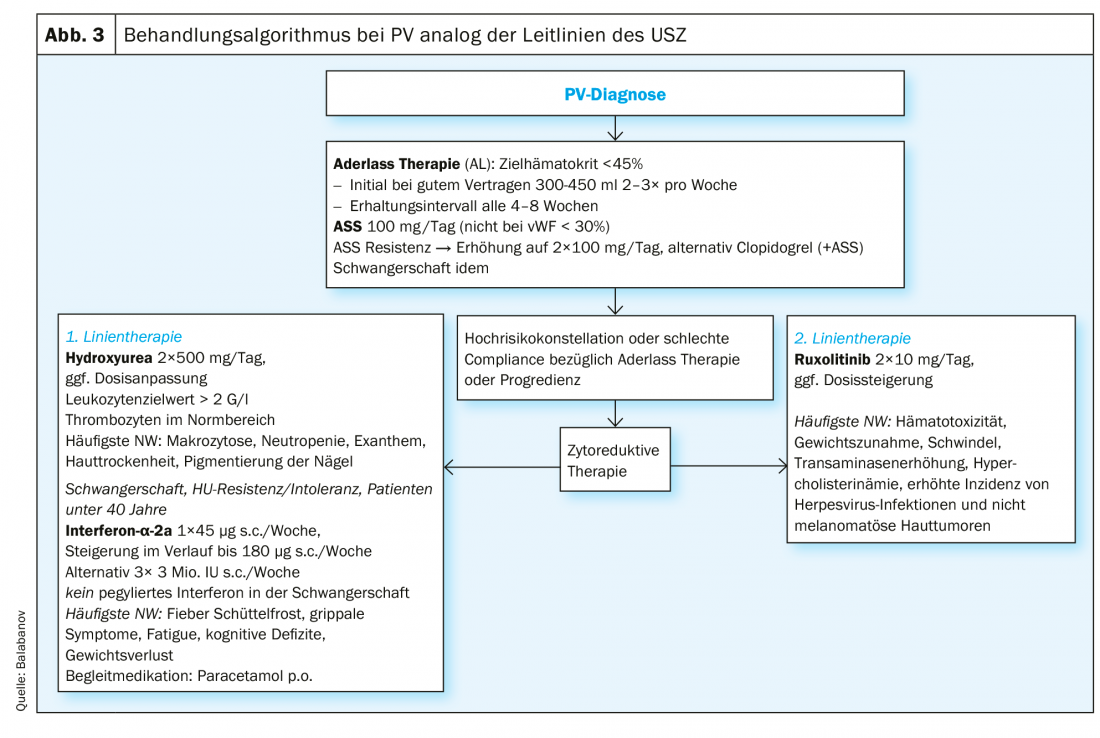
Patients à faible risque (âge <60 ans, antécédents négatifs d’événements thrombotiques) : Nous recommandons un traitement par saignées systématique, en visant un hématocrite cible <45%, car cette valeur cible est supérieure à des valeurs plus élevées (45-50%) en termes de survie sans événement [31]. En revanche, aucun effet positif sur les symptômes de la maladie n’a pu être documenté à ce jour [34]. Il est également recommandé de suivre un traitement anti-agrégant à base d’aspirine (ASS) pour prévenir les thrombus veineux et artériels [31–33]. Le traitement par ASA semble avoir un impact positif sur la symptomatologie associée en réduisant les troubles microvasculaires [35]. En cas de symptômes résistants à l’aspirine, on peut envisager d’administrer le clopidogrel deux fois par jour ou de l’utiliser seul ou en association avec l’ASA [36, 37]. Le cas échéant, un test de la fonction plaquettaire (agrégométrie plaquettaire, PFA-100) est indiqué pour distinguer les déficits congénitaux des causes iatrogènes [38]. La prudence est de mise lors de l’utilisation de l’aspirine, compte tenu de la possibilité d’une tendance accrue aux saignements chez les patients PV présentant une thrombocytose extrême concomitante (>1000×109/L).
les patients à haut risque (âge >60 ans, antécédents positifs d’événements thrombotiques) : Outre les mesures thérapeutiques susmentionnées, nous recommandons également un traitement cytoréducteur dans ce groupe de patients, l’objectif étant en premier lieu de normaliser l’hématocrite. Cependant, il faut également s’efforcer de réduire la thrombocytose et la leucocytose qui l’accompagnent souvent. Les recommandations actuelles concernant les substances pour le traitement de première ligne selon l’European LeukemiaNET (ENL) sont l’hydroxyurée et l’interféron α (INFα) (forme pégylée) [39]. Un échec thérapeutique sous hydroxyurée (tab. 2), au sens d’une résistance ou d’une intolérance, survient chez environ 24% des patients [7]. Le busulfan a été utilisé historiquement avec de bons résultats dans le traitement de deuxième ligne. Aujourd’hui, son utilisation est pour l’essentiel obsolète en raison d’un potentiel leucémogène suspecté, bien que l’on ne dispose pas de données solides sur l’augmentation de l’incidence des leucémies sous busulfan [40].

Le ruxolitinib, un inhibiteur de JAK2, est approuvé comme traitement de deuxième ligne de la PV et a montré des résultats positifs, avec une bonne tolérance, sur la myéloprolifération, la nécessité de saignées, ainsi que sur les symptômes associés à la PV tels que la fatigue et le prurit [41–44]. En cas de prurit réfractaire, un bénéfice a été décrit grâce à l’utilisation off-label des ISRS [45], INFα [46] et de la photothérapie [47]. Un résumé de l’algorithme de traitement de l’Hôpital universitaire de Zurich (USZ) est présenté dans la figure 3. Une greffe de moelle osseuse allogénique ou une greffe de cellules souches du sang périphérique ne doit être envisagée comme approche curative que dans des cas exceptionnels, chez des patients jeunes dont l’évolution est riche en complications ou dont la progression de la maladie ne peut être contrôlée par d’autres mesures [48]. En raison de l’augmentation du risque cardiovasculaire, tous les patients doivent être encouragés à réduire leur risque cardiovasculaire dans le cadre d’une prévention primaire (activité sportive, contrôle du poids, alimentation).
Grossesse : la PV n’est pas une contre-indication à la grossesse, mais elle est toujours considérée comme une grossesse à risque avec un taux d’avortement spontané plus élevé que dans la population normale [49]. En principe, nous recommandons une prise en charge dans un centre spécialisé disposant d’une expertise hématologique appropriée. Les recommandations relatives au traitement par saignées et au traitement antiagrégant sont essentiellement les mêmes que celles mentionnées ci-dessus. L’aspirine semble réduire le taux d’avortements spontanés et de complications de la grossesse [50]. En raison de l’absence de preuve de tératogénicité ou d’influence sur le taux de naissances vivantes [51] avec l’IFNα, un traitement est généralement possible et particulièrement recommandé pour les grossesses à haut risque [52].
Messages Take-Home
- La polycytémie vera est une néoplasie myéloproliférative des cellules souches hématopoïétiques, principalement causée par des mutations somatiques du gène JAK2 et l’hématopoïèse autonome clonale qui en résulte.
- Cliniquement, les symptômes et les complications mirco- et macrovasculaires sont liés à l’érythrocytose et à la thrombocytose qui l’accompagne souvent. Le risque de myélofibrose et de transformation leucémique est également important pour le pronostic.
- L’algorithme de diagnostic comprend des paramètres cliniques et de laboratoire, mais surtout la détection d’une mutation JAK2 dans la moelle osseuse, alors que d’autres aberrations génétiques prennent de plus en plus d’importance.
- Sur le plan thérapeutique, les patients sont répartis en groupes de risque. Alors que la thérapie par saignée et les inhibiteurs de l’agrégation plaquettaire sont utilisés indépendamment du risque, la thérapie cytoréductrice est principalement indiquée chez les patients à haut risque.
- Les grossesses chez les patientes PV sont toujours considérées comme des grossesses à risque et doivent être prises en charge de manière multidisciplinaire dans des centres spécialisés.
Littérature :
- Arber DA, et al : La révision 2016 de la classification de l’Organisation mondiale de la santé des néoplasmes myéloïdes et des leucémies aiguës. Blood, 2016. 127(20) : 2391-2405.
- Barbui T, et al : The 2016 revision of WHO classification of myeloproliferative neoplasms : Clinical and molecular advances. Blood Rev, 2016. 30(6) : 453-459.
- Jamieson CH, et al : La mutation JAK2 V617F se produit dans les cellules souches hématopoïétiques dans la polycythemia vera et prédispose à la différenciation érythroïde. Proc Natl Acad Sci U S A, 2006. 103(16) : 6224-6229.
- Bento C, et al : Base génétique de l’érythrocytose congénitale : mise à jour des mutations et bases de données en ligne. Hum Mutat, 2014. 35(1) : 15-26.
- Polycythemia vera : l’histoire naturelle de 1213 patients suivis pendant 20 ans. Gruppo Italiano Studio Policitemia. Ann Intern Med, 1995. 123(9) : 656-664.
- Moulard O, et al. : Epidémiologie de la myélofibrose, de la thrombocythémie essentielle et de la polycythémie vraie dans l’Union européenne. Eur J Haematol, 2014. 92(4) : 289-297.
- Tefferi A, et al : Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood, 2014. 124(16) : 2507-2513 ; quiz 2615.
- Crisa E, et al : A retrospective study on 226 polycythemia vera patients : impact of median hematocrit value on clinical outcomes and survival improvement with anti-thrombotic prophylaxis and non-alkylating drugs. Ann Hematol, 2010. 89(7) : 691-699.
- Barbui T., et al : La survie et la progression de la maladie dans la thrombocythémie essentielle sont significativement influencées par un diagnostic morphologique précis : une étude internationale. J Clin Oncol, 2011. 29(23) : 3179-3184.
- Tefferi A, et al : Survival and prognosis among 1545 patients with contemporary polycythemia vera : an international study. Leucémie, 2013. 27(9) : 1874-1881.
- Koschmieder S, et al : Myeloproliferative neoplasms and inflammation : whether to target the malignant clone or the inflammatory process or both. Leukemia, 2016. 30(5) : 1018-1024.
- Kroll MH, Michaelis LC, Verstovsek S : Mécanismes de la thrombogénèse dans la polycythémie vera. Blood Rev, 2015. 29(4) : 215-221.
- Finazzi G, Barbui T : Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leucémie, 2008. 22(8) : 1494-1502.
- Mesa RA, et al : The burden of fatigue and quality of life in myeloproliferative disorders (MPDs) : an international Internet-based survey of 1179 MPD patients. Cancer, 2007. 109(1) : 68-76.
- Saini KS, Patnaik MM, Tefferi A : Polycythemia vera-associated prurit and its management. Eur J Clin Invest, 2010. 40(9) : 828-834.
- Michiels JJ : Erythromelalgia and vascular complications in polycythemia vera. Semin Thromb Hemost, 1997. 23(5) : 441-454.
- De Stefano V, et al : Thrombose veineuse splanchnique et néoplasmes myéloprolifératifs : diagnostic à l’aide de molécules et traitement à long terme. Thromb Haemost, 2016. 115(2) : 240-249.
- Dentali F, et al : Thrombose veineuse cérébrale et néoplasmes myéloprolifératifs : résultats de deux grandes bases de données. Thromb Res, 2014. 134(1) : 41-43.
- Elliott MA, Tefferi A : Thrombosis and haemorrhage in polycythaemia vera and essential thrombocythaemia. Br J Haematol, 2005. 128(3) : 275-290.
- Michiels JJ, et al : The paradox of platelet activation and impaired function : platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost, 2006. 32(6) : 589-604.
- Pardanani A, et al : Prévalence et corrélations clinicopathologiques des mutations de l’exon 12 de JAK2 dans la polycythémie vera JAK2V617F-négative. Leukemia, 2007. 21(9) : 1960-1963.
- Vannucchi AM, et al : Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms : a critical reappraisal. Leukemia, 2008. 22(7) : 1299-1307.
- Passamonti F, et al : A prospective study of 338 patients with polycythemia vera : the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia, 2010. 24(9) : 1574-1579.
- Ward AC, Touw I, Yoshimura A : La voie Jak-Stat dans l’hématopoïèse normale et perturbée. Blood, 2000. 95(1) : 19-29.
- James C, et al. : Une mutation clonale unique de JAK2 conduisant à une signalisation constitutive provoque la polycythémie vera. Nature, 2005. 434(7037) : 1144-1148.
- Baxter EJ, et al : Mutation acquise de la tyrosine kinase JAK2 dans les troubles myéloprolifératifs humains. Lancet, 2005. 365(9464) : 1054-1061.
- Gangat N, et al : Études cytogénétiques au diagnostic dans la polycythémie vera : corrélations cliniques et de charge allélique JAK2V617F. Eur J Haematol, 2008. 80(3) : 197-200.
- Tefferi A, et al : Ciblage du séquençage profond dans la polycythémie vraie et la thrombocythémie essentielle. Blood Adv, 2016. 1(1) : 21-30.
- Scherber R, et al. : The Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF) : international prospective validation and reliability trial in 402 patients. Blood, 2011. 118(2) : 401-408.
- Grinfeld J, et al : Classification et pronostic personnalisé dans les néoplasmes myéloprolifératifs. N Engl J Med, 2018. 379(15) : 1416-1430.
- Marchioli R, et al : Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med, 2013. 368(1) : 22-33.
- Landolfi R, et al : Efficacité et sécurité de l’aspirine à faible dose dans la polycythémie vera. N Engl J Med, 2004. 350(2) : 114-124.
- Alvarez-Larran A, et al : Observation versus traitement antiplaquettaire comme prophylaxie primaire de la thrombose dans la thrombocythémie essentielle à faible risque. Blood, 2010. 116(8) : 1205-1210 ; quiz 1387.
- Grunwald MR, et al. : Caractéristiques cliniques et pathologiques de REVEAL au moment de l’inscription (ligne de base) : Étude prospective observationnelle de patients atteints de polycythémie veineuse aux États-Unis. Clin Lymphoma Myeloma Leuk, 2018. 18(12) : 788-795 e2.
- Michiels JJ, et al : érythromélalgiques cérébraux, oculaires et coronariens à médiation par les plaquettes.
InFo ONKOLOGIE & HÉMATOLOGIE 2019 ; 7(2-3) : 12-15