Les porphyries sont un groupe de maladies métaboliques très rares, d’origine génétique, caractérisées par un trouble de la biosynthèse de l’hème. La triade de symptômes typique d’une poussée est composée de douleurs abdominales, de symptômes cérébraux et d’une neuropathie périphérique. Un autre signe important est la coloration rouge de l’urine. Pour le traitement d’une poussée aiguë, l’administration intraveineuse d’hémarginate (Normosang®) est le traitement de choix. Lors de la prise en charge des patients, il est particulièrement important d’éviter les médicaments qui déclenchent des poussées (carte d’urgence). Les patients dont la porphyrie vient d’être diagnostiquée devraient être inclus dans le registre de Munich des porphyries aiguës.
Les porphyries sont un groupe de maladies métaboliques génétiques très rares qui sont dues à un trouble de la biosynthèse de l’hème. Dans l’organisme humain, l’hème est un composant de l’hémoglobine, responsable du transport de l’oxygène, et de la myoglobine, protéine de stockage de l’oxygène, dans lesquelles il est présent en tant que groupe prosthétique porteur de fer, ainsi que de nombreux cytochromes qui participent au métabolisme énergétique dans pratiquement toutes les cellules au niveau de la chaîne respiratoire . L’hémoglobine doit être fournie en permanence pour assurer le renouvellement continu de ces protéines.
Huit enzymes sont impliquées dans la biosynthèse de l’hème ; la régulation de la synthèse s’effectue par l’inhibition par rétroaction de la δ-ALA synthase, la première enzyme de la biosynthèse, par le produit final, l’hème. Il existe deux isoenzymes dans l’organisme : la δ-ALA synthase 1 est présente dans tous les tissus, la δ-ALA synthase 2 uniquement dans la moelle osseuse. En raison de leur importance quantitative, le foie et la moelle osseuse sont les principaux sites de synthèse de l’hème.
Physiopathologie des porphyries
Des mutations héréditaires (ou, plus rarement, spontanées) dans les gènes des enzymes de l’hémibiosynthèse entraînent une perte partielle de la fonction de l’enzyme codée. Cette perte partielle de fonction (en général à 50%) est suffisamment compensée par une stimulation des δ-ALA synthases, de sorte qu’il n’y a normalement pas de déficit en hème. Les porteurs de la prédisposition génétique n’ont donc généralement pas de problème avec la réduction de la performance métabolique de l’enzyme concernée. La situation devient problématique lorsque les besoins en hème augmentent, par exemple en raison de la prise d’un médicament comme la phénytoïne, qui induit des enzymes de dégradation du cytochrome P450 contenant de l’hème dans le foie. La synthèse de l’hème est alors très fortement stimulée et un “embouteillage” se produit au niveau du point d’étranglement du défaut enzymatique en raison de la réduction de l’activité à 50% : Les produits intermédiaires du métabolisme de l’hémoglobine (principalement les précurseurs de porphyrine, le porphobilinogène et le lévulinate de δ-amino) s’accumulent dans les cellules, passent dans l’espace extracellulaire et se dispersent dans l’organisme.
Trois formes de porphyrie
Selon l’enzyme affectée dans les porphyries aiguës, on distingue la variante fréquente “porphyrie aiguë intermittente” (PIA) et les variantes plus rares “porphyria variegata” (PV) et “coproporphyrinurie héréditaire” (HKP) (Fig. 1).
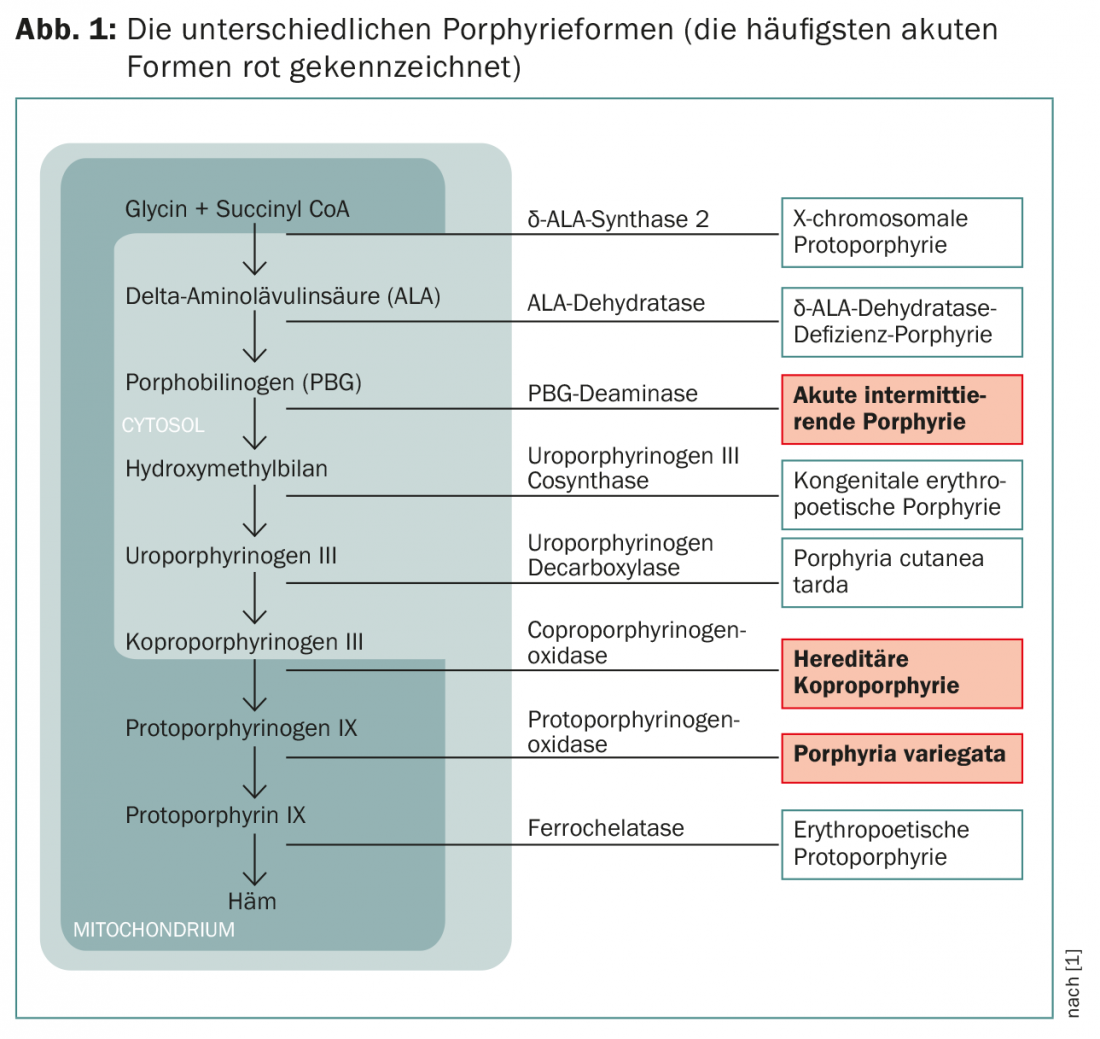
Les symptômes dépendent des métabolites qui s’accumulent. Les précurseurs de porphyrines encore hydrosolubles pénètrent dans les nerfs et provoquent des symptômes neurologiques, tandis que les porphyrines liposolubles se déposent dans la peau et provoquent des symptômes cutanés en cas d’exposition aux rayons UV. Ces troubles sont très variés et souvent déroutants, car ils sont également présents dans un grand nombre d’autres maladies. Le diagnostic correct est donc généralement posé tardivement.
Symptômes : douleurs abdominales, urine rouge, troubles neurologiques
Dans ces trois formes, l’apparition aiguë de symptômes est appelée poussée. Les poussées peuvent être légères et sont alors principalement déterminées par des douleurs abdominales. Ils sont récurrents, sans qu’il soit possible de poser le bon diagnostic. Ce n’est qu’en cas de poussée sévère, où les douleurs abdominales sont accompagnées de signes de symptômes centraux (tels qu’humeur dépressive, hallucinations, coma) et de symptômes neurologiques périphériques (douleurs dorsales, paralysie) (triade de symptômes), que la probabilité d’établir le bon diagnostic augmente (fig. 2).
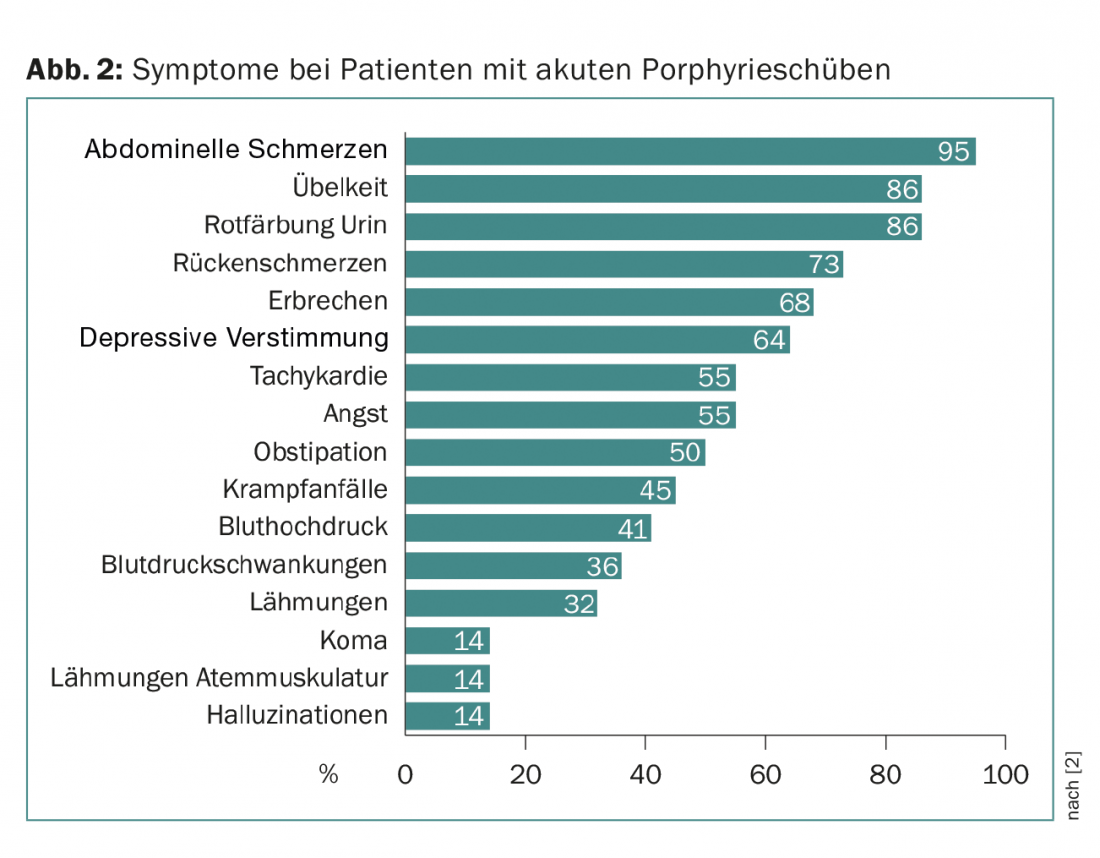
Un signe clinique supplémentaire important est la coloration rouge de l’urine, due à l’auto-oxydation du porphobilinogène en porphobiline. Associés à des modifications de laboratoire telles qu’une hyponatrémie, ces symptômes font suspecter une porphyrie aiguë (Fig. 3). Souvent, les patients sont pris en charge dans des unités de soins intensifs en raison de la gravité des symptômes, et c’est souvent un(e) jeune assistant(e) qui se souvient de cette maladie rare en raison de la proximité de l’examen d’État.
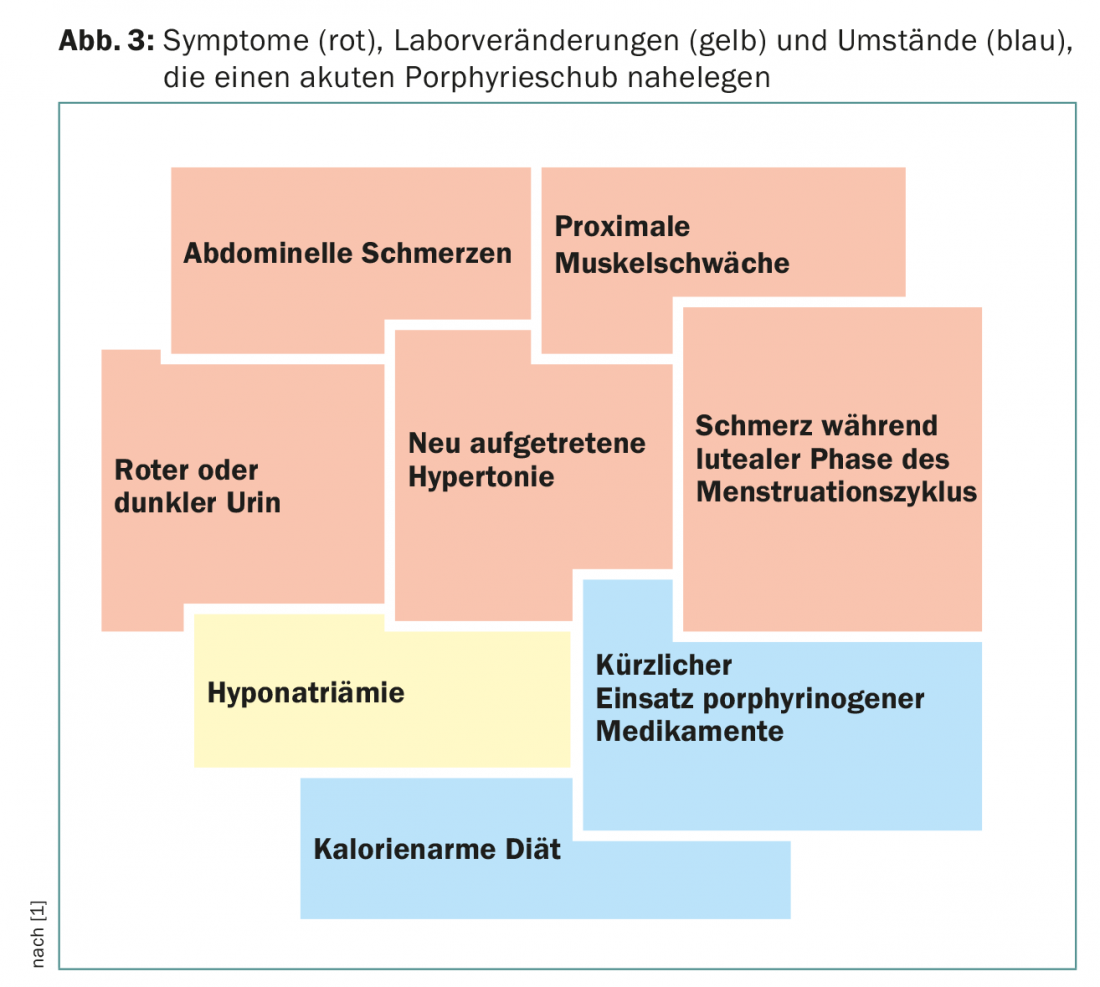
L’hyponatrémie peut entraîner des crises convulsives, qu’il n’est pas rare de traiter, par ignorance du diagnostic, avec de la carbamazépine ou de la phénytoïne, qui font partie des médicaments favorisant les poussées. Les symptômes cérébraux peuvent aller d’une légère altération psychique à une psychose paranoïaque, au délire ou au coma. Dans certains cas, on observe des mono- ou hémiparésies centrales ; en cas de paralysie respiratoire, la nécessité d’une ventilation mécanique menace, ce qui peut mettre la vie du patient en danger. Dans le cadre du syndrome d’encéphalopathie postérieure réversible (PRES), mis en évidence par un examen IRM, une cécité corticale avec perte totale de la vision peut se produire.
Une partie des patients développent une neuropathie périphérique, principalement motrice, au cours de la poussée. Chez une grande partie des personnes concernées, la polyneuropathie est déclenchée par des mesures iatrogènes (administration de médicaments porphyrinogènes) dans l’ignorance de la présence d’une porphyrie aiguë. La neuropathie est répartie de manière atypique, c’est-à-dire que les bras sont plus touchés que les membres inférieurs, les muscles proximaux sont plus marqués que les groupes musculaires distaux. Chez un tiers des patients, la paralysie débute au niveau des jambes, avec une accentuation proximale chez la moitié d’entre eux. Une distribution asymétrique des parésies est fréquente. Les signes d’une neuropathie sensorielle sont des troubles de la sensibilité soit en forme de chaussettes et de gants, soit au niveau du tronc comme un “maillot de bain”. Dans les porphyries aiguës plus rares, la PV et la HKP, des symptômes cutanés sont également présents. Cela est dû au fait que non seulement les précurseurs de porphyrine sont élevés, mais qu’en plus, en raison de la position du défaut enzymatique responsable, des produits intermédiaires ultérieurs de la synthèse de la porphyrine se déposent dans la peau.
Diagnostic
La détection accrue de précurseurs de porphyrines (δ-ALA et PBG) dans l’urine et non pas, comme on le croit encore souvent à tort, de porphyrines en premier lieu, permet de suspecter une porphyrie aiguë. Le porphobilinogène est d’abord mesuré de manière semi-quantitative dans un test rapide. Le problème est que ce test n’est pas disponible dans de nombreux laboratoires. Si le test est positif, l’analyse quantitative de la PBG et de la δ-ALA suit : si le résultat est rapporté à la créatinine urinaire, un échantillon d’urine spontané est généralement suffisant. L’urine de 24 heures n’est plus nécessaire.
Si les précurseurs de la porphyrine sont élevés, la présence d’une porphyrie aiguë intermittente est confirmée par le dosage de la PBG-désaminase dans les érythrocytes et, si celle-ci est abaissée, par une analyse mutationnelle (test génétique). Pour le diagnostic des deux autres porphyries aiguës, il est nécessaire d’analyser l’urine et les selles pour détecter respectivement les uroporphyrines et les coproporphyrines.
La confirmation finale du diagnostic se fait aujourd’hui par plus de 300 mutations différentes dans le gène de la PBG désaminase, ce qui signifie que pratiquement une position d’acide aminé sur deux peut être affectée par une mutation. C’est pourquoi le gène complet doit toujours être séquencé lors du premier diagnostic. En cas de mutation connue, il est possible de tester spécifiquement les proches pour détecter la présence de cette mutation.
Seuls 10 à 15% des porteurs du gène développent une manifestation de la porphyrie au cours de leur vie. Cela s’explique d’une part par le fait que la confrontation avec les agents déclencheurs n’a pas lieu lorsque l’on connaît la prédisposition et d’autre part par le fait que les gènes modificateurs protègent contre la manifestation d’une poussée.
Options thérapeutiques
Auparavant, le seul traitement disponible était la perfusion de glucose à haute dose, qui inhibe la synthèse de l’hème en régulant à la baisse l’activité de la δ-ALA-synthase. Aujourd’hui, le traitement de choix est l’administration intraveineuse d’hémarginate (Normosang®), qui permet de “freiner” l’hémiosynthèse. Le médicament est administré en perfusion courte sur quatre jours. Il est conseillé de le mélanger avec de l’albumine humaine en raison de son effet irritant sur les veines. Certains patients souffrant de poussées chroniques récurrentes reçoivent les perfusions via un port-a-cath. Il est clair que les médicaments favorisant les poussées doivent être arrêtés et ne doivent pas être réutilisés. Les médicaments classiques qui déclenchent des poussées sont par exemple les anticonvulsivants phénytoïne, carbamazépine, oxcarbazépine et acide valproïque, les barbituriques, les analgésiques métamizole et phénylbutazone, les antibiotiques sulfonamides et l’antifongique griséovulvine. Les médicaments considérés comme sûrs sont par exemple le lorazépam, le diazépam, la morphine, la péthidine, la chlorpromazine et l’ondansétron.
Les patients reçoivent une carte d’urgence, qui mentionne également le site Internet www.drugs-porphyria.com, où les listes de médicaments favorisant les poussées et de médicaments sûrs sont constamment mises à jour. Dans certains cas, il est difficile de procéder à une attribution sûre.
Prise en charge interdisciplinaire
Le diagnostic et le traitement des patients atteints de porphyries aiguës sont aujourd’hui réalisés par une équipe interdisciplinaire comprenant des hématologues, des gastroentérologues, des neurologues, des anesthésistes, des médecins de laboratoire, des dermatologues, des gynécologues et des généticiens [1]. Les aspects suivants sont particulièrement importants :
- Poussées chroniques récurrentes
- Traitement des complications neurologiques graves liées aux poussées
- Préparation et réalisation d’anesthésies
- Éviter les erreurs de diagnostic
- Prise en charge des patientes enceintes atteintes de porphyrie
- Diagnostic différentiel d’éventuelles modifications de la peau
- Fiabilité de l’analyse des mutations.
Nouveau diagnostic : inscription au registre de la porphyrie
En raison de la rareté de ces maladies, seuls les registres permettent d’obtenir des informations sur l’évolution, la sévérité des manifestations cliniques et l’altération de la qualité de vie, les éventuels effets secondaires des traitements, etc. Le registre munichois des porphyries aiguës a documenté 60 patients atteints de porphyries aiguës jusqu’en février 2016. Les patients nouvellement diagnostiqués doivent être signalés à ce registre [2]. Un site Internet à ce sujet est en cours de construction (www.akuteporphyrie.de).
Littérature :
- Petrides PE (éd.), et al. : Les porphyries aiguës, une brochure d’information pour les médecins. 4e édition 2015 Munich (disponible auprès d’Orphan Europe Ulm).
- Bronisch O, et al. : Acute Intermittent Porphyria in Germany : Interim Analysis of 45 Patients from a single institution (Munich). Congrès international sur les porphyrines et les porphyries, Düsseldorf, 9/2015.
InFo ONKOLOGIE & HÄMATOLOGIE 2016 ; 4(2) : 16-19