Les gliomes de grade II ont tendance à être sous-estimés dans leur malignité au stade précoce. Il n’existe pas de norme pour le traitement. Outre l’attentisme, une tactique chirurgicale agressive est de plus en plus souvent adoptée. Il n’est toutefois pas possible d’obtenir une guérison avec cette méthode.
Les gliomes de faible malignité de grade II de l’OMS infiltrent souvent le cortex et provoquent donc des crises d’épilepsie dans 60 à 80% des cas, souvent comme première manifestation [1,2]. C’est le lobe le plus grand du cerveau, le cerveau frontal, qui est le plus souvent touché. Il en résulte des changements de personnalité, des troubles de l’impulsion et de l’humeur, mais aussi une ataxie frontale. Si la région fronto pré-centrale à post-centrale est touchée, des crises focales motrices persistantes avec tendance à la généralisation se produisent, mais aussi des hémiparésies spastiques et des troubles de la sensibilité. En cas d’atteinte du lobe temporal, les crises partielles et complexes, voire les troubles du langage, dominent. Les troubles de l’appareil visuel sont rares, le plus probable étant l’hémianopsie homonyme en cas d’atteinte du tractus optique. Si le tronc cérébral est touché, il en résulte des troubles neurologiques complexes des voies longues et de la fonction des nerfs crâniens, pouvant aller jusqu’à la paralysie bulbaire avec troubles de la déglutition et de l’aspiration. Si le thalamus et les ganglions de la base sont atteints, des troubles extrapyramidaux ou des troubles de la mémoire apparaissent. Les fluctuations de la vigilance pouvant aller jusqu’à la démence sont au premier plan. Mais souvent, même en cas de processus très importants, la personnalité est bien préservée pendant une période prolongée, car les cellules tumorales envahissent le tissu cérébral sain de manière diffuse sans le détruire. Le lent déplacement des zones fonctionnelles par le tissu tumoral permet un déplacement de la fonction menacée vers les zones voisines ou du côté opposé (plasticité), ce qui est visible sur l’IRM fonctionnelle.
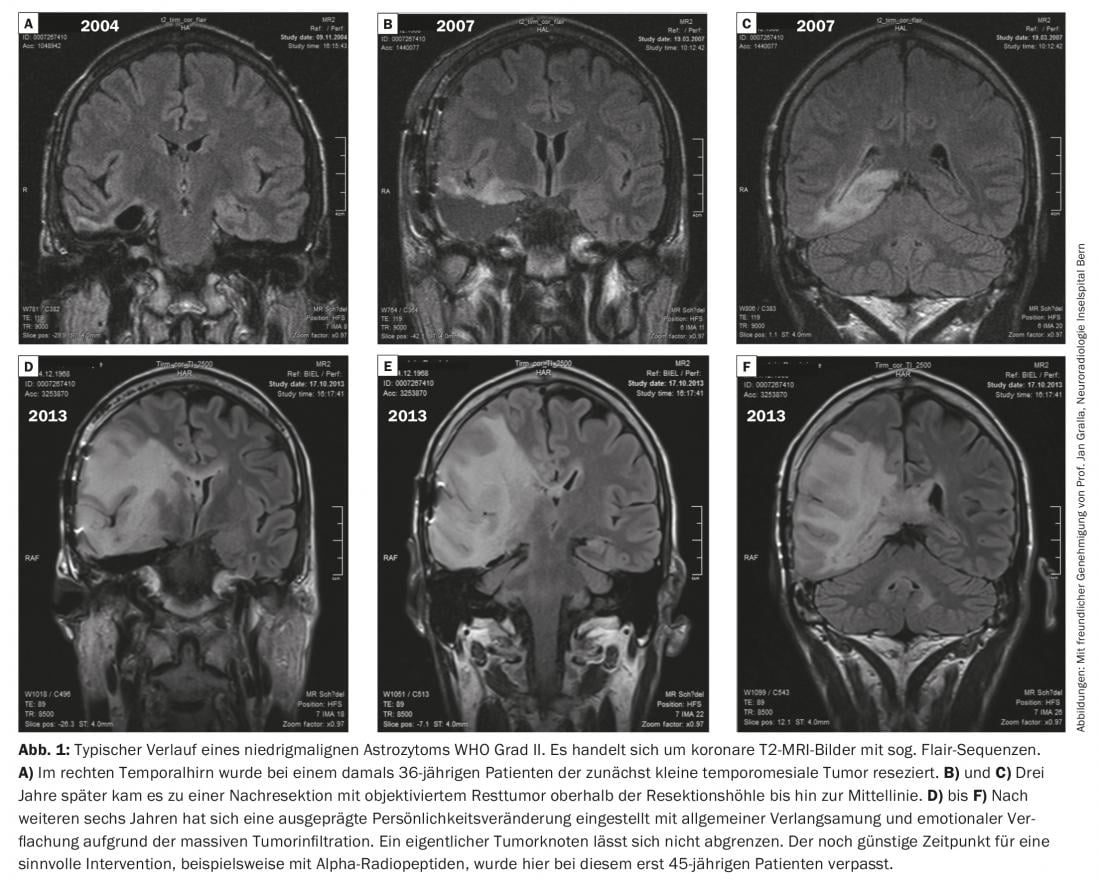
La figure 1 montre une évolution typique d’un astrocytome de faible malignité de grade II de l’OMS.
Les gliomes : Manifestation du taux de mutation spontanée ?
Rarement (<5%), les tumeurs cérébrales apparaissent de manière syndromique dans les cancers familiaux, par exemple dans le syndrome de Turcot avec défaut génétique “mismatch repair” ou dans le syndrome de Li-Fraumeni avec mutation p53 [3–5]. Plus rares encore sont les astrocytomes bénins à cellules géantes, qui se manifestent chez l’enfant et de manière sporadique chez l’adulte, et qui sont la conséquence d’une mutation congénitale des gènes de la sclérose tubéreuse TSC1 et TSC2, qui corégulent le complexe mTOR du métabolisme énergétique. L’anomalie génétique la plus fréquemment associée aux maladies neurologiques est la mutation du gène de la neurofibromatose de type 1, avec une fréquence de 1 pour 3000. Il s’agit de microdélétions intragéniques qui, dans la moitié des cas, ne sont pas héréditaires mais apparaissent spontanément. Le locus du gène NF1 sur le chromosome 17q11.2 est relativement instable. Le phénotype Nf1 comprend les gliomes optiques bénins. Des délétions de Nf1 ont été détectées dans des sous-types de glioblastomes et contribuent à la gliomagenèse en combinaison avec d’autres mutations. Dans les astrocytomes de grade II et dans le glioblastome secondaire, dont le pronostic est meilleur, on trouve souvent des mutations des gènes de l’isocitrate déshydrogénase IDH1 et IDH2. Comme la co-délétion 1p-19q dans l’oligodendrogliome, la mutation IDH marque un autre type d’origine biologique avec un meilleur pronostic.
Le taux de mutation spontanée est de 1:100’000 par division cellulaire. Un organisme passe par environ 1014 divisions cellulaires avant d’atteindre la différenciation complète. Au cours de la vie, d’innombrables erreurs de lecture se produisent dans chaque cellule lors de la division cellulaire – y compris dans le réservoir de cellules souches – qui sont pour la plupart immédiatement corrigées par des mécanismes de réparation intrinsèques à la cellule. Néanmoins, il y a toujours des mutations qui ne sont pas détectées ou qui ne sont pas prises en compte. ne sont pas réparées, ce qui contribue dans de rares cas au développement de tumeurs. Les gliomes font partie des maladies dites “orphelines”, c’est-à-dire très rares, avec une incidence de moins de cinq cas pour 10 000 personnes par an.
Classification génétique moléculaire
Des tests génétiques récemment développés, qui balayent l’ensemble du génome, permettent un pronostic assez précis de tous les types de gliomes et permettent de classer correctement les cas histologiquement incertains [6]. Il s’agit de balayer l’ensemble du génome à la recherche de profils de méthylation pathologiques. L’expression de nombreux gènes qui contrôlent la prolifération des cellules tumorales et favorisent l’apoptose est régulée par ce que l’on appelle les îlots CpG dans la région promotrice des gènes, qui peuvent être désactivés par méthylation, par exemple dans la différenciation des organes, mais aussi dans les cellules cancéreuses. Un autre type d’inactivation génique est la délétion à long terme de bras chromosomiques, qui transforme une section hétérozygote en section monozygote, souvent avec une réduplication. On peut alors créer un allélotype sur tous les chromosomes afin de détecter la perte d’hétérozygotie. Par exemple, les monosomies 1p et 19q sont pathognomoniques pour les oligodendrogliomes à croissance lente. Les oligodendrogliomes de type sauvage avec hétérozygotie 1p/19q ont un comportement beaucoup plus agressif. Dans les glioblastomes, on trouve typiquement la monosomie du chromosome 10 et la trisomie du chromosome 7 avec amplification de l’EGFR. L’analyse combinée de l’allélotype et du méthylome permet une classification très précise de tous les gliomes, en particulier dans les cas où l’histologie soulève des questions. Si des survivants à long terme sont observés dans des études sur le glioblastome, le diagnostic histologique devrait être vérifié par des tests moléculaires, car le taux d’erreur histologique dans les études à grande échelle peut atteindre 7% [7].
Réduction de masse retardée lors de la manifestation des symptômes
Comme la plupart des patients gliome de grade II sont en bon état clinique au moment du diagnostic, une phase d’observation est souvent choisie initialement [1,2]. De nombreux patients peuvent ainsi mener une vie normale relativement peu symptomatique pendant quelques années. Dans le cadre de cette stratégie, on n’intervient qu’en cas de transformation en un degré de malignité plus élevé (degré III ou IV) ou en cas d’échec de l’intervention. si des symptômes neurologiques se manifestent jusqu’à des signes de pression intracrânienne (céphalées, troubles de la vigilance, nausées à vomissements). Une réduction de masse est alors mise en place, suivie d’une radiochimiothérapie, ou résignée.
Crâniotomie éveillée et neuromonitoring
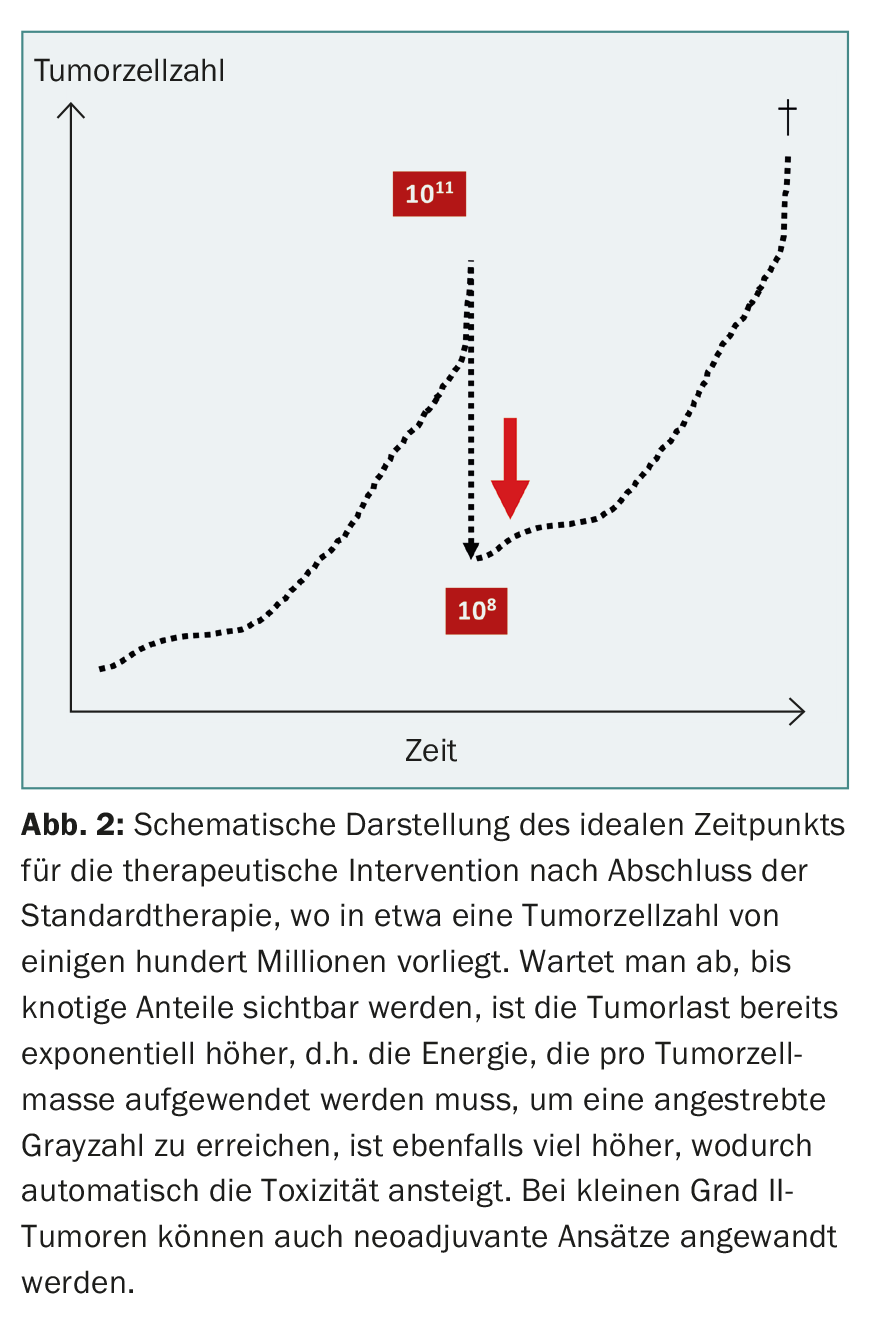
Dans les premières phases de la chirurgie du gliome, on a également tenté, dans des cas désespérés, de contrôler la tumeur par hémisphérectomie, mais cela n’a pas pu aboutir en raison de la tendance à l’infiltration diffuse des cellules tumorales. Ces dernières années, la chirurgie agressive des gliomes a connu une renaissance, mais en se limitant à une réduction de masse extensive par craniotomie à l’état de veille et neuromonitoring [8]. La réorganisation corticale due à la plasticité cérébrale permet parfois de réséquer des zones fonctionnellement importantes sans entraîner de déficits supplémentaires. Cette approche ne peut logiquement pas non plus aboutir à une guérison, car des millions de cellules tumorales infiltrantes restent non détectées. Un modèle de calcul permet de le démontrer : Un corps humain de 70 kg est composé d’environ 1014 cellules. Une tumeur de 70 g contient donc environ 1011 cellules tumorales. Lorsqu’une tumeur maligne infiltrante est réséquée à 99,9%, il reste encore environ 100 millions de cellules tumorales qui déterminent le sort futur (fig. 2). Un traitement moderne doit éliminer ces cellules résiduelles ou les remplacer par d’autres. contrôler.
Perspectives thérapeutiques
De nombreux patients atteints d’astrocytome de grade II n’ont pas dépassé l’âge de 50 ans ou ont été diagnostiqués à un stade précoce. seulement avec des déficits neurologiques croissants et sont exposés à la menace permanente d’une probabilité de 50 % de transformation en un niveau de malignité supérieur. Cela plaide en fait pour une intervention précoce. Bien que la résection supramaximale n’entraîne pas de guérison, elle réduit le risque de transformation maligne. Les premières expériences d’irradiation ciblée de cellules individuelles par des biomolécules diffusibles qui se fixent sur des récepteurs spécifiques de cellules tumorales et qui portent comme effecteur un isotope de haute énergie à très courte portée ont conduit à plusieurs reprises à un contrôle très long de la tumeur, bien au-delà de la survie moyenne, sans toxicité importante [9,10]. D’autres médicaments viendront s’y ajouter à l’avenir, qui bloqueront les circuits biologiques perturbés et amélioreront ainsi le pronostic [5].
Messages Take-Home
- Environ 10 à 15% de tous les gliomes malins se présentent principalement sous la forme d’astrocytomes de grade II de faible malignité ou, plus rarement, d’oligodendrogliomes. La survie moyenne est de sept à dix ans pour les astrocytomes de grade II et de dix à quinze ans pour les oligodendrogliomes de grade II.
- Compte tenu du pronostic misérable des glioblastomes plus fréquents, les gliomes de grade II ont tendance à être sous-estimés dans leur malignité à la phase précoce.
- Les tumeurs cérébrales sont probablement la conséquence du taux de mutation spontanée des processus cellulaires, en quelque sorte le revers de la recombinaison.
- La nouvelle classification de l’OMS des tumeurs cérébrales sur la base du schéma de méthylation et de l’allélotype permet d’établir un pronostic fiable.
- Il n’existe pas de norme pour le traitement des gliomes de grade II. Outre l’attitude attentiste, une tactique chirurgicale agressive est de plus en plus souvent adoptée. Cependant, en raison de l’infiltration des cellules tumorales dans les tissus cérébraux sains, il n’est pas possible d’obtenir une guérison. Il existe de premières indications selon lesquelles la radiothérapie ciblée sur une seule cellule avec des biomolécules diffusibles pourrait améliorer considérablement le pronostic.
Littérature :
- Merlo A, de Tribolet N : Tumeurs du cerveau et de la moelle épinière. In : Steck A, Hess CH (éd.) : Neurologische Pathophysiologie. Berne : Éditions Hans Huber 2003.
- Schneider T, et al : Gliomes chez l’adulte. Dtsch Arztebl Int 2010 ; 107(45) : 799-807.
- Merlo A, Rochlitz C, Scott R : Survival of patients with Turcot’s syndrome and glioblastoma [letter]. N Engl J Med 1996 ; 334(11) : 736-737.
- Merlo A, Bettler B : Glioblastomas on the move. Science STKE 2004 ; 2004(229) : pe18.
- Lino M, Merlo A : Translating Biology into Clinic : the case of glioblastoma. Curr Opin Cell Biol 2009 ; 21(2) : 311-316.
- Louis DN, et al : The 2016 World Health Organization Classification of Tumors of the Central Nervous System : a summary. Acta Neuropathol 2016 ; 131(6) : 803-820.
- Linz U : Commentary on Effects of Radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study : 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466). Cancer 2010 ; 116(8) : 1844-1846.
- Duffau H : The Rationale to Perform Early Resection in Incidental Diffuse Low-Grade Glioma : Toward a “Preventive Surgical Neurooncology”. World Neurosurgery 2013 ; 80(5) : e115-e117.
- Cordier D, et al : Traitement ciblé par alpha-radionucléide des gliomes fonctionnellement critiqués avec la substance P de 213Bi-DOTAGA : un essai pilote. Eur J Nucl Med Mol Imaging 2010 ; 37(7) : 1335-1344.
- Cordier D, et al : Composés radiolabellisés ciblés dans le traitement du gliome. Semin Nucl Med 2016 ; 46(3) : 243-249.
InFo ONKOLOGIE & HÉMATOLOGIE 2017 ; 5(6) : 7-10