Un diagnostic sûr, des patients convaincus de l’utilité du traitement, cela peut paraître banal, mais ces aspects sont très pertinents dans la perspective d’un traitement médicamenteux qui pourrait durer toute la vie. Outre un aperçu concis des différentes substances actives, les particularités du traitement médicamenteux chez les enfants sont abordées.
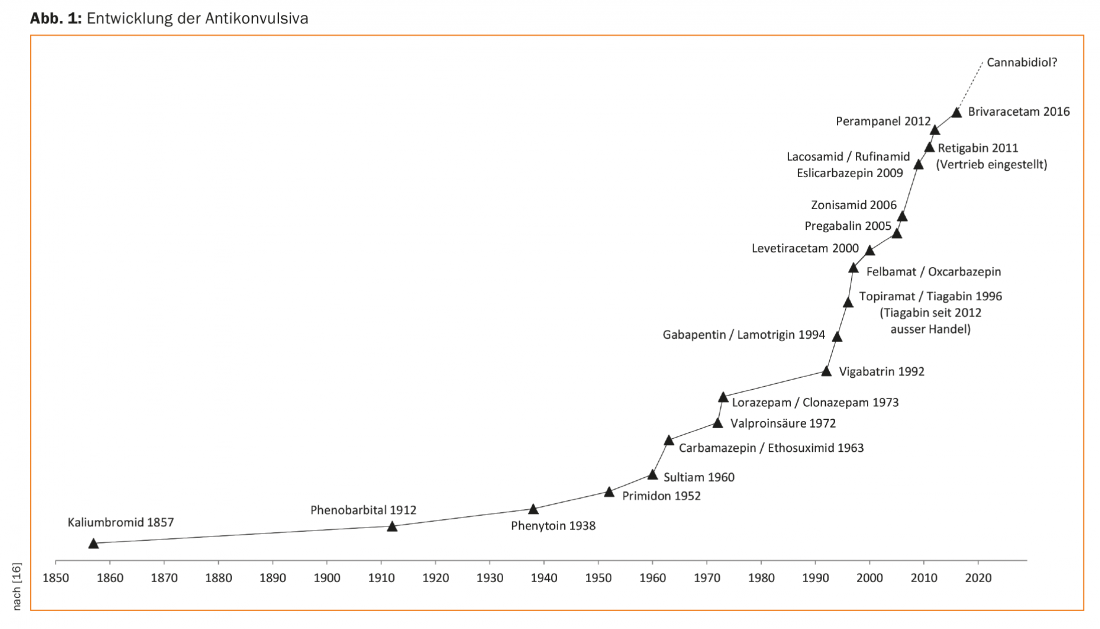
Les crises d’épilepsie et l’épilepsie sont des situations cliniques fréquentes et connues depuis des millénaires. Les tablettes cunéiformes de l’époque babylonienne du IIe siècle av. avant J.-C. les premiers textes médicaux sur l’épilepsie sont consignés. Au fil des siècles, de nombreuses méthodes différentes ont donc été utilisées pour traiter les crises d’épilepsie. Il est vrai qu’Hippocrate, vers 500 av. L’épilepsie était considérée comme une maladie d’origine cérébrale, mais cette connaissance a été oubliée et les patients atteints d’épilepsie ont donc longtemps été considérés (et le sont encore aujourd’hui dans certaines régions du monde) comme des personnes possédées par des démons et traitées par des exorcismes et autres mesures fondées sur la religion. Ce n’est qu’aux XVIIe et XVIIIe siècles qu’il a été possible d’obtenir une autorisation de construire. et surtout au 19e siècle, la vision actuelle des sciences naturelles s’est lentement imposée [1]. En 1857, le gynécologue de la reine Victoria, Sir Charles Locock, a décrit pour la première fois dans Lancet l’effet antiépileptique du bromure de potassium, fondant ainsi le traitement médicamenteux moderne de l’épilepsie [2]. En 1912, un autre médicament a été ajouté, le phénobarbital, qui joue encore un rôle dans le traitement de l’épilepsie aujourd’hui. Depuis lors, de nombreuses substances ont été découvertes ou sont en cours de découverte. (Fig. 1), bien qu’aucun effet antiépileptique n’ait été démontré à ce jour pour ces substances, mais seulement un effet anticonvulsivant, c’est-à-dire supprimant les crises. Il serait donc plus correct de parler d’anticonvulsivants et non d’antiépileptiques.
Pour pouvoir mettre en œuvre un bon traitement médicamenteux de l’épilepsie, il faut tout d’abord que le diagnostic d’épilepsie soit confirmé. Cela peut sembler banal, mais c’est loin de l’être dans la pratique clinique quotidienne, car souvent, surtout au début d’une épilepsie, on ne dispose que de descriptions rudimentaires des crises et le taux d’erreurs de diagnostic est étonnamment élevé. Les syncopes, en particulier, peuvent être accompagnées de manifestations cloniques dues à des troubles du rythme cardiaque ou à des dérégulations circulatoires, simulant ainsi une crise d’épilepsie tonico-clonique. Les troubles fonctionnels ou les troubles moteurs hyperkinétiques, tels que les myoclonies, doivent également être pris en compte dans le diagnostic différentiel. Étant donné que le traitement de l’épilepsie, en particulier chez les adultes, implique souvent une thérapie médicamenteuse à vie et requiert une forte adhésion au traitement, le patient doit être convaincu du bien-fondé du traitement. En cas d’incertitude, il convient donc d’envisager un examen vidéo EEG de longue durée à bas seuil pour confirmer le diagnostic.
Causes
Outre l’anamnèse et la description des crises, le diagnostic des épilepsies repose principalement sur l’EEG et l’imagerie, si possible une IRMc. Il faut également tenir compte ici des progrès techniques des appareils IRM, de sorte qu’en cas d’IRM négative, il peut être utile de répéter l’examen avec un appareil plus récent et de le réaliser avec des protocoles IRM spécifiques à l’épilepsie. Le diagnostic doit permettre de déterminer les causes ou les effets les plus précis possibles. la classification dans un syndrome épileptique peut être obtenue, ce qui influence le choix de l’anticonvulsivant approprié.
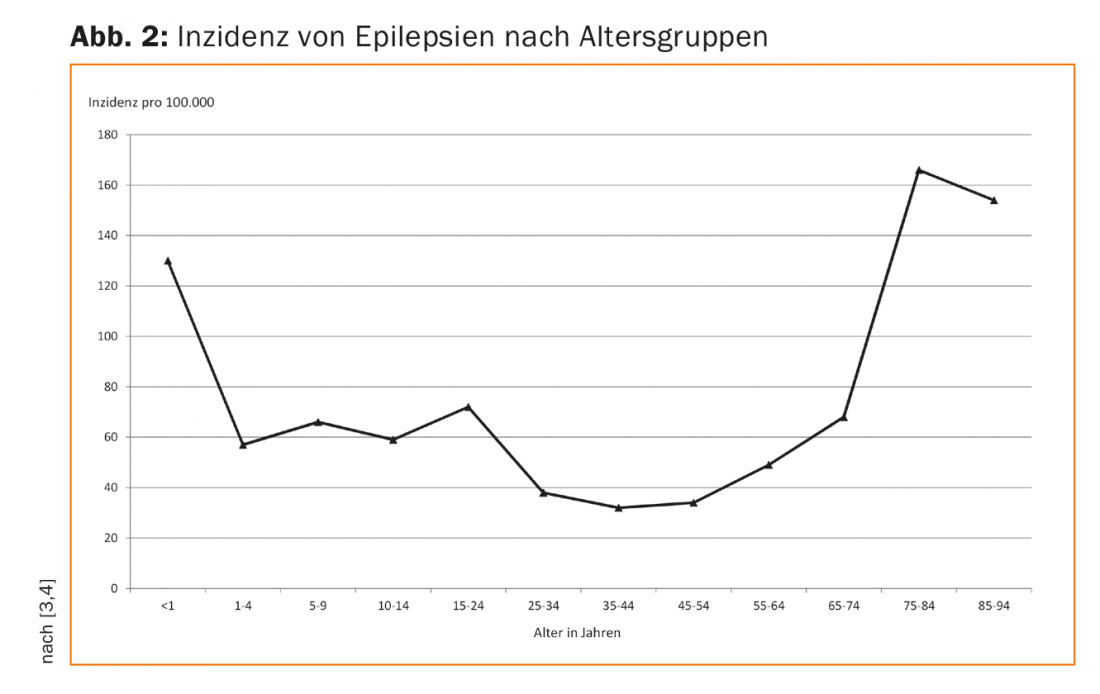
Par nature, les causes de l’épilepsie diffèrent en partie chez les enfants et les adultes. Ainsi, chez les enfants, les épilepsies dues à des prédispositions génétiques (comme l’épilepsie par absence et l’épilepsie de Rolando), à des malformations cérébrales congénitales ou à des asphyxies périnatales, ainsi que les crises dues à des troubles du métabolisme (comme les épilepsies dépendantes de la vitamine B6) prédominent. Les épilepsies dues à des lésions cérébrales acquises sont également fréquentes chez les enfants. Or, contrairement aux enfants, ces derniers représentent chez les adultes la grande majorité des épilepsies nouvellement survenues. En raison de l’évolution démographique, l’incidence de l’épilepsie chez les personnes âgées est en nette augmentation, ce qui pose de nouveaux défis en matière de traitement médicamenteux dans ce groupe d’âge (Fig. 2).
Épileptologie pédiatrique
Dans le cadre du diagnostic et du traitement de l’épilepsie chez les enfants, une dimension supplémentaire est le niveau de développement et l’âge des enfants. Ainsi, la fréquence et le type d’épilepsie diffèrent nettement selon les groupes d’âge pédiatriques concernés. Historiquement, il a longtemps été difficile de savoir comment classer les nombreux syndromes au sein du grand groupe des troubles épileptiques ; le développement du diagnostic EEG par Hans Berger au milieu du siècle dernier a permis de faire de grands progrès dans ce domaine. On peut citer à titre d’exemple l’épilepsie de Watanabe, le syndrome de West, l’épilepsie de Rolando et l’épilepsie par absences. L’épilepsie Watanabe, par exemple, concerne les crises bénignes du nouveau-né qui apparaissent au cours des trois premiers mois et disparaissent à la fin de la première année de vie. Le syndrome de West a été décrit pour la première fois par William James West en 1841 à Tonbridge, et il a présenté les crises caractéristiques de Nick Salam en un éclair. Le syndrome de West est une épilepsie généralisée qui s’accompagne de spasmes et se manifeste généralement entre trois et six mois. Il est déclenché par une multitude de causes possibles (polyétiologie). Le syndrome de West est à cet égard un bon exemple de la manière dont des causes différentes conduisent à une voie physiopathologique finale commune et présentent ainsi le même tableau clinique électroencéphalographique et sémiologique. L’épilepsie de Rolando, ou épilepsie focale idiopathique bénigne, est un autre trouble épileptique pédiatrique classique. Elle a également un âge typique et survient généralement chez les jeunes enfants et les jeunes écoliers. Le début de la maladie est caractérisé par des paresthésies de fourmillements et des manifestations motrices au niveau du visage, et souvent par un arrêt de la parole. L’EEG montre alors des “sharp-waves” triphasiques classiques au-dessus de la région centrale. Chez les enfants un peu plus âgés, on observe l’épilepsie-absences juvénile classique, qui s’accompagne d’une persistance et d’une absence de réactivité à la sollicitation. Le diagnostic de suspicion est souvent posé par les enseignants, car les enfants semblent absents en classe en raison de leur “rêverie”. L’EEG montre alors des paroxysmes généralisés de 3/s spike-wave pendant les absences.
Cet extrait des syndromes épileptiques pédiatriques laisse déjà entrevoir que le traitement doit être spécifiquement mis en place après caractérisation du syndrome épileptique [5].
Thérapie médicamenteuse chez les enfants
Le choix de l’anticonvulsivant pour le traitement continu chez les enfants est déterminé de manière décisive par la caractérisation du trouble épileptique. Outre le syndrome épileptique, d’autres aspects tels que le développement et l’objectif thérapeutique sont bien entendu déterminants pour le choix. Cependant, pour toutes les épilepsies, le traitement aigu d’une crise repose sur une benzodiazépine, généralement le midazolam ou le clonazépam. Pour les soins ambulatoires, il existe des formes orales, rectales et nasales de midazolam.
Pour les épilepsies watanabe et les épilepsies bénignes du nourrisson, la carbamazépine ou l’oxcarbazépine se sont imposées. Le syndrome de West est classiquement traité selon un schéma thérapeutique comprenant de la vigabatrine et des stéroïdes. L’épilepsie de Roland est classiquement traitée en première intention par le sultiam ou l’oxcarbazépine, tandis que l’épilepsie par absences est généralement traitée par l’éthosuximide.
Outre ces syndromes épileptiques classiques, il existe bien entendu chez les enfants des épilepsies qui sont la conséquence de modifications structurelles, par exemple après un accident ou une infection. Dans ce cas, la lamotrigine, le lévétiracétam ou l’oxcarbazépine se sont imposés, comme à l’âge adulte. Dans de nombreux autres syndromes épileptiques, comme l’épilepsie myoclonique juvénile ou le syndrome de Lennox-Gastaut, ainsi que l’épilepsie myoclonique-astatique, le valproate s’est imposé comme le traitement de première intention [7].
En règle générale, en épileptologie pédiatrique, il est préférable d’opter pour une monothérapie afin d’éviter tout effet négatif sur le développement de l’enfant [6]. En ce qui concerne les objectifs du traitement, il est important de tenir compte du fait que le degré de mobilité de l’enfant a une influence décisive sur l’objectif du traitement. Ainsi, un enfant qui se déplace dans la rue ou qui va à la piscine, par exemple, est nettement plus exposé aux crises qu’un enfant immobile. En outre, avec l’âge et l’indépendance croissante vis-à-vis des parents, d’autres aspects tels que la prise autonome de la médication et l’observance du traitement sont au premier plan.
Dans de nombreux syndromes épileptiques, un nombre non négligeable de patients ne feront plus de crises à la fin de la maturation cérébrale, même sans médicament, de sorte que l’épileptologue pédiatrique doit planifier une tentative d’arrêt. Il convient donc de discuter suffisamment tôt avec la famille de la date et de la manière dont une tentative de sortie pourrait avoir lieu, même si elle se situe dans plusieurs années. En outre, pour les patients qui auront besoin d’anticonvulsivants toute leur vie, il faut veiller à ce que les soins soient continus lors du passage de la pédiatrie à la médecine adulte.
Traitement médicamenteux chez l’adulte
Une fois le diagnostic d’épilepsie confirmé, il convient de discuter avec le patient du symptôme cible à traiter. Dans la plupart des cas, il s’agira de “grandes” crises tonico-cloniques propagées de manière bilatérale (“grand-mal”), qui représentent un stress considérable pour les personnes concernées, mais aussi bien sûr pour leurs proches, et qui comportent un risque de blessure pouvant aller jusqu’à la mort subite inattendue (SUDEP – “sudden unexpected death in epilepsy patients”). Même les crises purement focales peuvent parfois évoluer vers une grande crise tonico-clonique bilatérale, avec les conséquences que cela implique. Certains patients remarquent ou ne se souviennent pas de leurs crises, ce qui rend souvent difficile la notion d'”absence de crises” dans la pratique clinique quotidienne. Crises d’épilepsie répétées une fréquence élevée des crises (y compris des crises infracliniques) peut également entraîner des troubles cognitifs au cours de l’évolution [8]. L’indication du traitement doit donc être discutée avec le patient au cas par cas.
Le traitement commence alors par un anticonvulsivant approprié (voir ci-dessous), qui est dosé à une première dose cible. Si d’autres crises surviennent sous cette dose, il faut d’abord épuiser la dose de ce produit jusqu’au seuil de tolérance avant de passer à une autre substance ou d’établir un traitement combiné (add-on). Le seuil de tolérance varie considérablement d’une substance à l’autre et d’un individu à l’autre.
De nombreuses substances sont désormais disponibles pour le traitement de l’épilepsie chez l’adulte. Les substances les plus utilisées aujourd’hui sont la lamotrigine, le lévétiracétam, le valproate et la carbamazépine, et de nouvelles préparations sont régulièrement mises sur le marché. Toutefois, les nouvelles substances ne sont pas nécessairement plus efficaces que les anciennes, mais elles ont beaucoup moins d’effets secondaires et généralement moins d’interactions avec d’autres médicaments [7]. Néanmoins, dans les grandes études sur les médicaments, qui sont généralement menées sur des patients épileptiques réfractaires, un certain nombre de patients (environ 10-20%) ne font même plus de crises avec une nouvelle substance, ce qui peut justifier un essai thérapeutique correspondant avec une nouvelle substance.
Lamotrigine : cette préparation est disponible depuis le milieu des années 1990 et est généralement très bien tolérée, sans effets secondaires. Il peut être utilisé aussi bien pour les épilepsies focales (structurelles) que pour les épilepsies généralisées. Il s’agit d’un bloqueur des canaux sodiques disponible uniquement par voie orale. Son principal inconvénient est qu’il doit être administré très lentement pour éviter les redoutables réactions allergiques cutanées qui peuvent aller jusqu’au syndrome de Lyell ou de Stevens-Johnson, potentiellement mortel. La première dose cible habituelle est de 150 mg (pour les personnes âgées) et 200 mg (dose standard) et n’est atteinte qu’après 4 à 6 semaines. En raison de sa longue demi-vie d’environ 24 à 35 heures, la lamotrigine peut être administrée une seule fois par jour. Si nécessaire, la dose peut souvent être considérablement augmentée au cours de l’évolution en raison de sa bonne tolérance, jusqu’à 500-800 mg par jour [9].
L’utilisation pendant la grossesse est possible et n’est associée qu’à une faible augmentation du risque de malformation [10]. Dans la vie quotidienne, il faut tenir compte de certaines interactions médicamenteuses importantes de la lamotrigine : ainsi, l’éthinylestradiol contenu dans les contraceptifs hormonaux contenant des œstrogènes et des progestatifs (“pilule”) peut pratiquement diviser par deux le taux plasmatique de la lamotrigine, ce qui peut conduire d’une part à une protection insuffisante contre les crises pendant la prise de la pilule et d’autre part à des taux nettement plus élevés pendant l’interruption de la prise de la pilule, avec les effets secondaires correspondants. [11]. Les femmes doivent donc être conseillées en conséquence et nous recommandons, en cas de traitement par la lamotrigine, soit un progestatif pur, soit un stérilet. Dans le cas des thérapies combinées, il faut surtout tenir compte de l’interaction avec le valproate. Le valproate inhibe considérablement la dégradation de la lamotrigine, de sorte qu’il faut choisir un schéma de dosage progressif beaucoup plus lent et une dose totale de lamotrigine plus faible [9]. D’autre part, cette combinaison est souvent très efficace en raison de la lenteur du métabolisme et donc des faibles variations des taux.
Lévétiracétam : ce produit peut être utilisé dans les épilepsies focales mais aussi généralisées. Il est disponible sous forme de comprimés, de sirop et par voie intraveineuse, ce qui lui confère un rôle important dans le traitement des crises aiguës à l’hôpital. La première dose cible est de 750 mg (personnes âgées) à 1000 mg (dose standard). Il peut être augmenté jusqu’à environ 4000 mg et est administré deux fois par jour (demi-vie d’environ 7 heures). L’effet est médié par la protéine vésiculaire pré-synaptique SV2. Le lévétiracétam est peu lié aux protéines plasmatiques et n’a pratiquement aucun potentiel d’interaction. Cependant, les principaux effets secondaires peuvent être des symptômes psychiatriques tels que l’irritabilité, l’agitation interne, l’agressivité, l’anxiété et la dépression. Dans la littérature, le taux de ces effets secondaires est estimé à 10-15%, mais en pratique clinique quotidienne, même si cela ne conduit pas toujours à un changement de traitement, ce taux semble être encore plus élevé, de l’ordre de 20-30%. Son utilisation chez les patients présentant des troubles du comportement et/ou, par exemple, des lésions cérébrales frontales, n’est donc pas favorable. Le lévétiracétam a un bon effet sur les myoclonies. Avec la lamotrigine, il s’agit d’un médicament de premier choix en cas de grossesse.
Le brivaracétam (Briviact®) est une évolution du lévétiracétam, le dernier anticonvulsivant à avoir été autorisé en Suisse fin 2016. Cette préparation a moins d’effets secondaires psychiatriques que le lévétiracétam avec une efficacité (au moins) égale, la dose cible est de 100-200 mg, répartie en deux doses, elle peut être administrée rapidement resp. il est possible de commencer directement avec la dose cible. Lors du passage du lévétiracétam au brivaracétam (par exemple en raison de symptômes psychiatriques), un changement dans un rapport de 10-15 à 1 peut être effectué “du jour au lendemain” (par exemple, 1000 mg de lévétiracétam à 100 mg de brivaracétam).
Valproate : Le valproate est une substance découverte dans les années 1960 qui joue toujours un rôle important dans le traitement de l’épilepsie. Il agit probablement en renforçant les mécanismes GABAergiques et est utilisé dans les épilepsies focales ainsi que dans les épilepsies généralisées. Pour ces derniers en particulier, il est souvent considéré comme le traitement le plus efficace. La première dose cible habituelle est de 1000 mg, répartie en deux doses, la préparation à libération prolongée pouvant n’être administrée qu’une fois par jour. Contrairement à la plupart des autres anticonvulsivants, le valproate est principalement métabolisé par voie hépatique, ce qui permet de l’utiliser même en cas d’insuffisance rénale. Les effets secondaires les plus pertinents sont une prise de poids souvent importante et un éventuel ralentissement cognitif. Le valproate est un puissant inhibiteur enzymatique, de sorte que les interactions pertinentes doivent être prises en compte lors du traitement dans la pratique clinique quotidienne. En cas d’association avec la lamotrigine, la dégradation de la lamotrigine est inhibée, ce qui augmente significativement le taux de lamotrigine et peut donc entraîner des signes d’intoxication (voir ci-dessus). Un traitement antibiotique par carbapénème peut entraîner une chute importante des taux de valproate dans les 24 à 48 heures, pouvant aller jusqu’au déclenchement d’un état de mal épileptique [12]. Le métabolisme dans le foie peut entraîner une augmentation de la production d’ammoniaque, ce qui peut provoquer une encéphalopathie. Pour les femmes en âge de procréer, il est conseillé de prendre un traitement de substitution. ne doit pas être utilisé en raison d’une tératogénicité importante. ne doit être utilisé qu’après une évaluation détaillée du rapport bénéfice/risque et une déclaration de consentement écrite de la femme concernée (voir également la section Grossesse).
Carbamazépine : la carbamazépine a également été découverte dans les années 1960. Il s’agit d’un bloqueur des canaux sodiques utilisé dans les épilepsies focales. Les épilepsies généralisées peuvent entraîner une aggravation des crises, notamment des myoclonies et des absences. La première dose cible habituelle est de 800 mg, répartie en deux doses (demi-vie due à l’auto-induction : 16-24 heures ; en cas d’administration unique : environ 36 heures [9]). L’effet secondaire le plus pertinent de la carbamazépine est le vertige, parfois accompagné d’un nystagmus, en raison de sa faible marge thérapeutique. Les réactions cutanées ne sont pas non plus rares avec la carbamazépine, ce qui est associé à des allèles HLA spécifiques, présents surtout chez les Japonais, les Chinois et les Thaïlandais, mais aussi, un peu plus rarement, chez les Caucasiens. Comme la carbamazépine est un inducteur enzymatique puissant et qu’il existe donc de nombreuses interactions médicamenteuses, son utilisation est souvent limitée chez les patients âgés, bien qu’il existe en soi un bon effet anticonvulsivant. Il peut être utilisé pendant la grossesse, même si les taux de malformations sont légèrement plus élevés qu’avec la lamotrigine et le lévétiracétam [10].
Situations particulières – Grossesse
Les jeunes femmes en âge de procréer souffrant d’épilepsie ont un besoin particulier de conseils en vue d’une éventuelle grossesse. L’épilepsie en soi n’est pas une raison pour ne pas tomber enceinte et dans plus de 90% des cas, les femmes atteintes d’épilepsie donnent naissance à des enfants en bonne santé et sans complications. Il y a cependant quelques éléments importants à prendre en compte [13]. Une substitution en acide folique doit être commencée dès le désir d’enfant, c’est-à-dire avant même le début de la grossesse (voir également l’article sur l’épilepsie et le désir d’enfant en page 18 de ce numéro).
Le plus important est certainement de trouver le meilleur réglage médicamenteux pour protéger la femme et le bébé à naître des crises pendant la grossesse. Le premier choix chez les femmes enceintes est la lamotrigine et le lévétiracétam, qui n’ont montré qu’une faible augmentation du risque de malformation dans de grandes études prospectives de registre (par exemple, le registre EURAP) [10]. Contrairement à la lamotrigine et au lévétiracétam, le risque de malformation est très élevé avec le valproate (jusqu’à environ 45%( !) en fonction de la dose, surtout des anomalies du tube neural, mais aussi des troubles cognitifs chez l’enfant [14]). L’effet tératogène se manifeste surtout au cours des premières semaines de la grossesse, lorsque la femme ne sait souvent même pas qu’elle est enceinte, de sorte que le valproate ne doit pas être utilisé chez les femmes en âge de procréer ; des avertissements correspondants (“lettre rouge”) ont été émis par les autorités de réglementation. Il serait souhaitable d’inclure toutes les femmes atteintes d’épilepsie et suivant un traitement anticonvulsivant dans un registre de grossesse tel que le registre EURAP mentionné ci-dessus, afin d’obtenir des informations encore plus sûres sur la tératogénicité des différentes substances. Les formulaires correspondants peuvent être téléchargés sur Internet et remplis par chaque médecin et envoyés à un centre d’étude approprié.
Limites de la thérapie – Chirurgie de l’épilepsie
Bien que l’absence de crises soit souvent l’objectif principal du traitement, cet objectif n’est atteint en moyenne que chez 60 à 70% des patients, selon le syndrome épileptique. Même les thérapies combinées ne permettent qu’à un petit nombre de patients de ne plus avoir de crises. Ces taux n’ont pas augmenté de manière significative au cours des dernières décennies, même avec les nouveaux médicaments. Outre la vérification du diagnostic d’épilepsie et de l’observance, il convient donc d’envisager rapidement une intervention chirurgicale de l’épilepsie. Cela peut conduire à une guérison durable de l’épilepsie chez les patients appropriés atteints d’épilepsie focale, jusqu’à environ 80-90% selon l’origine des crises. Ces dernières années, on a donc commencé à passer de la chirurgie de l’épilepsie en dernier recours à un traitement de (presque) première intention (par exemple en cas d’épilepsie du lobe temporal mésial due à une sclérose de l’hippocampe – voir également l’article sur le traitement chirurgical à la page 26 de ce numéro). [15]).
Si l’on sait que certaines épilepsies de l’enfant et de l’adolescent peuvent “guérir” de la maladie, c’est rarement le cas à l’âge adulte. Néanmoins, dans certaines circonstances, une tentative prudente d’arrêt peut être justifiée (erreur de diagnostic DD dans le passé ?). D’autre part, si la cause de l’épilepsie persiste (par exemple, une malformation, un défaut post-traumatique ou post-ischémique) et qu’il n’y a pas de crises, il ne faut pas la mettre en péril.
De manière pragmatique, nous diminuons la dose à une dose faible en cas d’épilepsie généralisée, d’absence de crises depuis de nombreuses années et de demande expresse du patient, tout en effectuant des contrôles EEG (l’EEG est plus utile pour prédire le risque de récidive des crises dans les épilepsies généralisées que dans les épilepsies focales). Nous effectuons ensuite la dernière étape du dosage sous surveillance vidéo-EEG à long terme, mais il reste bien sûr un certain risque résiduel de récidive que le patient doit être prêt à assumer.
Messages Take-Home
- Le diagnostic d’épilepsie doit être confirmé.
- La fréquence et le type d’épilepsie varient considérablement selon les groupes d’âge pédiatriques.
- Le choix de l’antiépileptique pour le traitement continu chez les enfants est déterminé de manière décisive par la caractérisation du trouble épileptique.
- Dans de nombreux syndromes épileptiques, les patients ne feront plus de crises une fois la maturation du cerveau terminée, même sans médicament, de sorte qu’une tentative d’arrêt doit également être planifiée à long terme.
- En épileptologie pédiatrique, une monothérapie doit être recherchée dans la mesure du possible.
- Les doses doivent être augmentées à partir de la première dose cible, en fonction des crises, jusqu’au seuil de tolérance ; ce n’est qu’ensuite que l’on passe à un autre produit ou à un add-on (traitement combiné/pour les adultes).
- Les interactions médicamenteuses doivent être prises en compte : La carbamazépine est un inducteur enzymatique, le valproate un inhibiteur enzymatique, les carbapénèmes peuvent réduire drastiquement les taux de valproate.
- Dans la mesure du possible, le valproate ne doit pas être utilisé chez les femmes en âge de procréer.
Littérature :
- Gonzalo A (éd.) : Introduction à l’épilepsie. Cambridge University Press ; 2012.
- Brody MJ : Traitement médicamenteux antiépileptique – l’histoire jusqu’à présent. Seizure 2010 Dec ; 19(10) : 650-655.
- Werhahn KJ : Epilepsy in the elderly (L’épilepsie chez les personnes âgées). Dtsch Arztebl Int. 2009 ; 106(9) : 135-142.
- Olafsson E, et al : Incidence des crises non provoquées et de l’épilepsie en Islande et évaluation de la classification du syndrome épileptique : une étude prospective. Lancet Neurol 2005 ; 4 : 627-634
- Broser P, Maier O : Encéphalopathies épileptiques infantiles précoces. Epileptologie 2016 ; 33 : 95-101.
- Pellock JM, Bourgeois BFD, Dodson WE (éd.) : Epilepsie pédiatrique. New York, Demos, 2007.
- Steinhoff B, Bast T : Vade-mecum antiépileptique. Société allemande d’épileptologie 2017.
- Vingerhoets G : Effets cognitifs des seizures. Seizure 2006 ; 15(4) : 221-226.
- Swissmedic : www.compendium.ch. Consulté en juillet 2018
- Tomson T, et al : Risque comparatif de malformations congénitales majeures avec huit médicaments antiépileptiques différents : une étude de cohorte prospective du registre EURAP. Lancet Neurol 2018 Jun ; 17(6) : 530-538.
- Sabers A, et al : Lamotrigine plasma levels reduced by oral contraceptives. Epilepsy Res 2001 Nov ; 47(1-2) : 151-144.
- Sutter R, Rüegg S, Tschudin-Sutter S : Seizures as adverse events of antibiotic drugs : A systematic review. Neurology 2015 Oct 13 ; 85(15) : 1332-13341.
- Voinescu PE, Penell PB : Prise en charge de l’épilepsie pendant la grossesse. Expert Rev. Neurother 2015 ; 15(10) : 1171-1187.
- Meador KJ et al : Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study) : a prospective observational study. Lancet Neurol 2013 Mar ; 12(3) : 244-252.
- Steinhoff BJ, Staack AM : Y a-t-il une place pour le traitement chirurgical de l’épilepsie non pharmacorésistante ? Epilepsy Behav 2018.
- Golyala A, Kwan P : Drug development for refractory epilepsy : The past 25 years and beyond ; Seizure 2017 Jan ; 44 : 147-156.
InFo NEUROLOGIE & PSYCHIATRIE 2018 ; 16(5) : 12-17