Le syndrome d’Ehlers-Danlos (EDS) est un groupe de troubles congénitaux de la synthèse du collagène. Il survient chez environ 1/5000 à 1/10 000 personnes et est à peu près aussi fréquent chez les deux sexes. Il existe 13 sous-types, dont l’expression peut parfois varier considérablement. La plupart d’entre elles suivent un mode de transmission autosomique dominant, mais certaines formes sont également transmises selon un mode autosomique récessif.
“Tout le monde la connaît, la douleur pointue, aiguë, chaude, sourde, stridente. Je les connais toutes, les différentes teintes de la douleur et les nuances angoissantes entre les deux. Elle m’accompagne à chaque respiration, pendant mon sommeil, quand je m’habille le matin, tout simplement tout le temps”. (Jasmin Polsini)*
Les différents symptômes du syndrome d’Ehlers-Danlos (EDS) sont dus à des mutations de différents gènes qui ont une influence sur le tissu conjonctif. Suite à ces mutations, la structure du tissu conjonctif est perturbée. Comme les mutations affectent différents gènes, les symptômes peuvent varier considérablement d’un patient à l’autre, ce qui rend le diagnostic du SDE difficile.
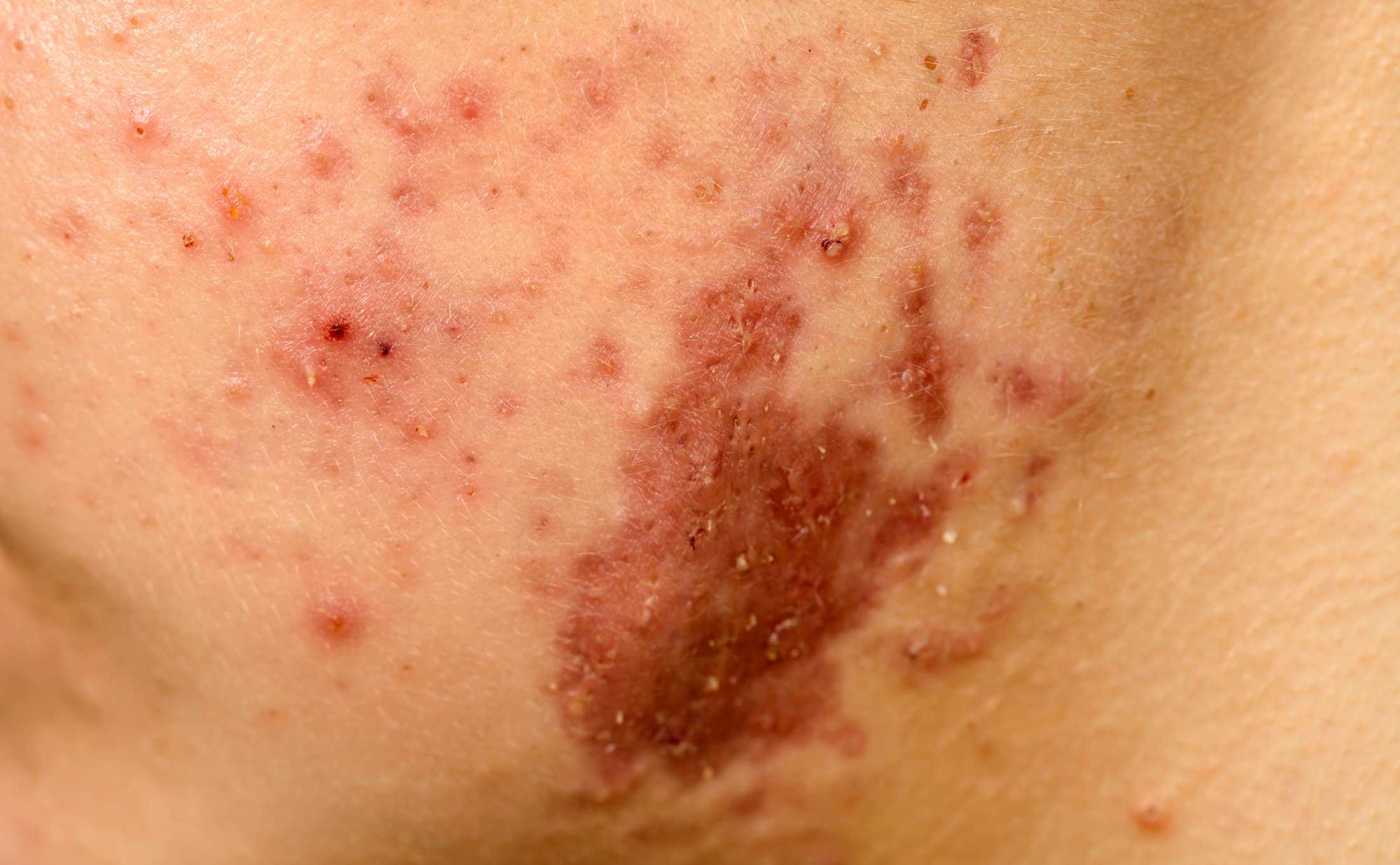
Le SDE présente un très large éventail de symptômes. Le SDE se caractérise par des articulations hyper-extensives avec des (sub)luxations fréquentes, une peau hyper-élastique et une fragilité des tissus.
Le tissu conjonctif des vaisseaux sanguins peut également être affecté. Cela entraîne un retard de cicatrisation et la formation fréquente d’hématomes. En outre, de gros vaisseaux sanguins peuvent être touchés, ce qui peut entraîner par exemple une dilatation de la racine aortique ou un prolapsus de la valve mitrale. Des hernies, des troubles gastro-intestinaux tels que la dyspepsie, le reflux gastro-œsophagien ou le syndrome du côlon irritable peuvent être détectés. La dysautonomie et la fatigue chronique sont plus fréquentes que la moyenne chez les patients EDS. Les états douloureux aigus et chroniques peuvent avoir les causes et les degrés les plus divers et sont très éprouvants sur le plan psychologique, ce qui peut entraîner des dysfonctionnements psychologiques tels que la dépression, l’anxiété ou les troubles affectifs.
Un diagnostic difficile
En raison de la variabilité des symptômes, il existe un grand nombre de diagnostics différentiels possibles. Les principaux symptômes qui affectent la peau et l’appareil locomoteur sont également présents dans de nombreuses maladies rhumatologiques. Le syndrome de Marfan et la cutis laxa présentent également des symptômes similaires. Souvent, les patients EDS ont également été mal diagnostiqués avec la fibromyalgie ou même le “syndrome de l’enfant battu”.
Si l’on soupçonne une maladie du tissu conjonctif, une anamnèse familiale détaillée est utile. Lors de l’examen clinique, l’hypermobilité des articulations est déterminée à l’aide du score d’hypermobilité de Beighton, ce qui peut déjà indiquer la possibilité d’un SDE. Il convient de noter que la mobilité dépend également d’autres facteurs tels que l’âge, l’état général et le sexe. La structure du collagène peut être déterminée au microscope électronique après une biopsie.
Certains types d’EDS peuvent être analysés par amplification de l’ADN suivie d’une analyse de séquence. L’important dans le diagnostic est d’exclure les autres causes possibles des symptômes, ce qui rend indispensable un diagnostic différentiel étendu.
Bien que le syndrome d’Ehlers-Danlos ne puisse pas être guéri, il peut être traité. Un diagnostic précoce est donc extrêmement important. En cas de suspicion de SDE, les nouveaux critères de diagnostic permettent de poser un diagnostic clinique et de le confirmer par un test génétique.

Le long chemin du syndrome de DE
Il n’existe pas encore de traitement causal pour l’EDS, ni de directives standardisées pour la gestion de l’EDS. Chez les patients atteints de SDE, l’accent est mis sur le traitement de la douleur et des symptômes multiples dans le cadre d’une approche multidisciplinaire. L’intervention chirurgicale n’est guère une option, car les patients EDS présentent des résultats chirurgicaux moins prévisibles, en plus d’un risque opératoire plus élevé.
Il peut souvent s’écouler jusqu’à 30 ans entre le début des symptômes et le diagnostic correct. De nombreux patients atteints de SDE se plaignent d’un délai trop long avant d’être diagnostiqués. Ils ont fait l’expérience que les médecins et les spécialistes avaient tendance à classer leurs symptômes dans la catégorie “psychosomatique”, qui ne nécessite pas d’examen plus approfondi, et à exclure en soi une maladie héréditaire en raison de sa rareté.
Auteurs :
Maximilian Sabev, Simon Schading, Florian Schneider, Jasmin Polsini, Zurich
Information pour le médecin et le patient :
https://ehlers-danlosnetzschweiz.blogspot.ch
*www.schlafendehundewecken.ch
PRATIQUE DU MÉDECIN DE FAMILLE 2018 ; 13(3) : 10