Le 55e congrès annuel de la “American Society of Hematology” s’est tenu fin 2013. Plus de 22 000 participants sont venus à la Nouvelle-Orléans ou “The Big Easy”, comme on l’appelle. Les personnes intéressées trouveront le programme complet avec des extraits des présentations sur www.hematology.org.
Transplantation haplo-identique de cellules souches
(mb) La transplantation de cellules souches hématopoïétiques HLA haploidentiques est une option efficace pour les patients qui ont besoin d’une transplantation, mais qui ne trouvent pas de donneur avec des types HLA totalement compatibles. C’est la conclusion du Dr Alice Bertaina, Hôpital pour enfants Bambino Gesu, Rome. Comparé à une greffe de cellules souches provenant d’un donneur entièrement compatible, un tel traitement est généralement associé à un risque plus élevé d’infection et de rechute. La méthode présentée par Bertaina consiste à éliminer sélectivement les cellules T alpha/bêta+ ainsi que les cellules B CD19+ du greffon du donneur. Parallèlement, on conserve des cellules immuno-actives comme les cellules tueuses naturelles et les cellules T gamma/delta+. Au total, 45 enfants atteints de leucémie aiguë – âgés de 0,9 à 17,9 ans au moment de la transplantation – ont ainsi été traités avec les cellules souches d’un parent (35 patients atteints de LAL et 10 de LAM). Le délai moyen pour atteindre un nombre absolu de neutrophiles de >0,5 × 109/l et un nombre de plaquettes de >50 × 109/l était de 13 (9-18) et 11 jours (8-20), respectivement. Aucun enfant n’a développé de réaction aiguë du greffon contre l’hôte (GVHD) dans l’intestin ou le foie. Une GVHD légère est apparue dans la peau chez 29% d’entre eux. Après un suivi de onze mois en moyenne (entre 2 et 30), l’estimation de Kaplan-Meier à deux ans s’élevait à un taux de survie sans leucémie de 75% (IC à 95% : 57-86). Ce chiffre était de 73% (IC 95% : 52-85) pour les patients atteints de LAL. “Nos résultats avec les cellules souches haploidentiques de donneurs ont montré des pourcentages de réussite identiques à ceux d’une transplantation avec correspondance complète”, a déclaré le Dr Bertaina. “Ainsi, ce traitement qui sauve des vies sera accessible à un nombre beaucoup plus important de patients sans correspondance parfaite avec le donneur”.
Récepteur antigénique chimérique (CAR)
Plusieurs chercheurs ont présenté une autre prouesse technique : une thérapie utilisant des lymphocytes T modifiés par CAR pour traiter des maladies oncohématologiques. Le récepteur antigénique chimérique (CAR) est une chaîne d’ADN construite artificiellement qui est insérée dans les cellules T d’un patient à l’aide de vecteurs (connus en génétique moléculaire). Ces “nouvelles” cellules combinent la spécificité des anticorps et l’action cytotoxique des lymphocytes T. Elles sont donc capables d’attaquer les cellules cancéreuses.
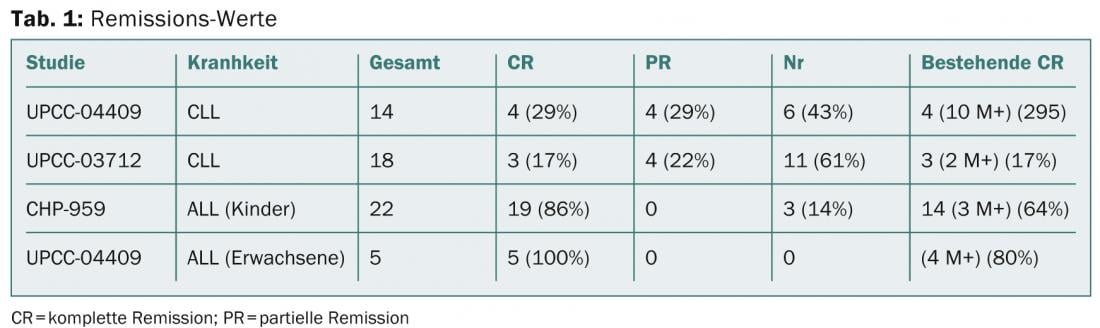
Michael Kalos de l’Université de Pennsylvanie Perelman School of Medicine à Philadelphie a présenté les résultats d’études sur la LLC et la LLA avancées, récidivantes ou réfractaires (Porter et al NEJM 2011 ; Kalos et al. Sci Trans Med 2011, Grupp et al. NEJM 2013, abstract 67). Au total, 32 patients atteints de LLC fortement prétraités ont été traités, dont huit ont présenté une rémission partielle et sept une rémission complète. Les résultats sont présentés dans le tableau 1.
CAR dans les lymphomes B
Le rapport de James Kochenderfer, MD, du National Cancer Institute du National Institute of Health à Bethesda, prouve que les patients atteints de lymphome médiastinal primaire à cellules B (PMBCL) réfractaire à la chimiothérapie et de lymphome diffus à grandes cellules B (DLBCL) peuvent également bénéficier du traitement CAR anti-CD19. Les chercheurs ont traité 20 patients avec 23 perfusions de cellules T au total. Les neuf premiers traitements de cellules CAR-T ont déjà été rapportés (Kochenderfer et al. dans Blood 2010 et Blood 2012). Lors de l’ASH, Kochenderfer a maintenant présenté les résultats non encore communiqués de 14 patients. La chimiothérapie précédente ayant clairement montré que l’activité des cellules T transmises par adoption s’améliorait, les patients reçoivent du cyclophosphamide et de la fludarabine pendant cinq jours pour une perfusion de cellules T CAR anti-CD19. Cinq patients ont obtenu une rémission complète (RC) et six une rémission partielle (RP).
“C’est le premier rapport sur un traitement réussi du PMBCL réfractaire à la chimiothérapie et du DLBCL avec des cellules T anti-CD19-CAR”, a déclaré Kochendorfer. “Nos données suggèrent pour la première fois le potentiel de cette approche pour les patients atteints de lymphomes agressifs, qui étaient jusqu’à présent presque impossibles à traiter”.
Comparaison de Zevalin et du rituximab
Les traitements de consolidation par Zevalin (ibritumomab-tiuxetan 90Y) et rituximab ont tous deux montré un bénéfice considérable en termes de survie sans progression (PFS). Une étude espagnole de phase II a montré que le traitement de consolidation par rituximab était supérieur au traitement de consolidation par zevalin chez les patients atteints de lymphome folliculaire et répondant au R-CHOP. De juin 2008 à juillet 2010, 146 patients (66 hommes et 80 femmes d’un âge moyen de 55 ans) provenant de 25 établissements espagnols ont été inclus. Le Dr Armando Lopez-Guillermo de l’Hospital Clinic de Barcelone a présenté les résultats préliminaires. Après un suivi de 37 mois en moyenne à partir de la randomisation (entre 26 et 56), 31 patients ont présenté une évolution avancée ou une récidive. La SSP à 36 mois était de 64% (IC à 95% : 52-76) pour les patients du groupe Zevalin et de 86% (IC à 95% : 77-95) pour les patients du groupe rituximab (p=0,01 ; HR 0,38, IC à 95% : 0,170,83). Pendant la période de maintien, six des 63 patients du groupe Zevalin ont présenté une neutropénie (grade 3-4) et cinq d’entre eux une thrombocytopénie (3-4). Pour les patients du groupe rituximab, c’était un ou aucun des 61 patients. Cinq patients sont décédés pendant les examens de suivi en raison de la progression du lymphome. Aucune différence n’a été constatée entre les groupes.
Le bortézomib dans le myélome multiple
Dans le myélome multiple (MM), l’induction par bortézomib plus traitement d’entretien continue d’améliorer la survie des patients symptomatiques nouvellement diagnostiqués. C’est le résultat de l’étude hOVON65, dont les résultats à long terme ont été présentés par le Dr Pieter Sonneveld du Erasmus Medisch Centrum, Rotterdam. Dans cette étude, 827 patients atteints de MM ont été randomisés après un traitement d’induction par VAD (vincristine, doxorubicine, dexaméthasone ; n=414) ou PAD (bortézomib, doxorubicine, dexaméthasone ; n=413), suivi d’une forte dose de melphalan et d’une autogreffe de cellules souches. Le traitement d’entretien consistait en 50 mg de thalidomide par jour (pour le groupe VAD) ou 1,3 mg de bortézomib/m2 iv toutes les deux semaines (pour le groupe PAD) pendant deux ans. La réponse pendant le traitement protocolaire semble s’être légèrement améliorée maintenant que tous les patients ont terminé le traitement : Une rémission complète (RC) plus une rémission complète nodulaire (RCN) a été observée dans 49% des cas avec la PAD et dans 35% des cas avec la VAD. Une très bonne rémission partielle (VGPR) a été observée chez 26% et 21% respectivement, et une ≥ rémission partielle (PR) chez 91% et 83% respectivement. Après un suivi de 67 mois en moyenne, 111 des patients traités par VAD et 131 des patients traités par PAD n’ont pas progressé et sont restés en vie. La survie sans progression (le temps écoulé entre la randomisation et la progression, la récidive ou le décès) s’est avérée meilleure avec le PAD (ajusté au stade) (ISS) (HR 0,78 ; IC à 95% 0,66-0,91 ; p=0,002). Le PAD était également supérieur pour l’objectif secondaire de survie globale après ajustement ISS (HR 0,80 ; IC à 95% 0,65-1,00 ; p=0,047).
Nouvelle mutation dans la TE et la PMF JAK-2-négatives
Des chercheurs autrichiens et italiens ont découvert une nouvelle mutation spécifique aux patients atteints de thrombocytémie essentielle (TE) JAK-2 négative et de myélofibrose primaire (PMF). L’altération génétique la plus fréquente dans le syndrome myéloprolifératif (MPN) est la mutation JAK2-V617F. Celle-ci survient chez 95% des patients atteints de polycythémie vera (PV) et chez 50 à 60% des patients atteints de TE et de PMF. Des mutations dans l’exon 12 de JAK2 et dans le gène du récepteur de la thrombopoïétine MPL sont observées dans 5 à 10 % des cas supplémentaires. Ces dernières années, il est apparu que plusieurs autres gènes étaient atteints dans le cas du MPN. Cependant, ces mutations sont également présentes dans d’autres maladies myéloïdes. Un marqueur moléculaire spécifique pour les 40% restants de patients ET et PMF avec JAK2 et MPL de type sauvage est donc le bienvenu.
Thorsten Klampfl, du Centre de recherche en médecine moléculaire de l’Académie autrichienne des sciences, à Vienne, et ses collègues ont utilisé le “whole exome sequencing” pour identifier de nouvelles mutations chez des patients atteints de PMF avec JAK2 et MPL de type sauvage. L’analyse a montré des insertions et des délétions somatiques récurrentes dans le codage CALR pour la calréticuline. Toutes les mutations détectées résultaient d’un décalage du cadre de lecture et étaient regroupées dans le dernier exon (exon 9) du gène. Suite à cette découverte, les chercheurs ont mis au point un test basé sur la PCR pour rechercher des mutations d’insertion/délétion dans l’exon 9 du CALR chez 1 107 patients atteints de MPN. Aucune mutation n’a été trouvée chez les patients PV, mais elle l’a été chez les patients ET et PMF : les mutations CALR étaient mutuellement exclusives avec JAK2 muté et MPL muté. Chez les patients atteints de JAK2 sauvage et de MPL, 67% des patients ET et 88% des patients PMF présentaient un CALR muté. Aucune mutation de l’exon 9 de CALR n’a été détectée chez 254 patients atteints de LAM de novo, chez 45 patients atteints de leucémie myéloïde chronique, chez 73 patients atteints du syndrome myélodysplasique ou chez 64 patients atteints de leucémie myéloïde chronique.
Les auteurs estiment que ce marqueur sera bientôt à la disposition des cliniciens pour améliorer les décisions diagnostiques et thérapeutiques dans le cas du NPP.
Source : 55e réunion annuelle de l’ASH, 7-10 décembre 2013, Nouvelle-Orléans
InFo Oncologie & Hématologie 2014 ; 2(3) : 30-32
Spécial Congrès 2014 ; 5(2) : 6-7