Si la formation de nouvelles cellules sanguines dans la moelle osseuse est perturbée, une forme rare de cancer du sang chronique peut en être la cause. L’hyperprolifération des trois rangées de cellules de la moelle osseuse entraîne une érythrocytose, une thrombocytose et une leucocytose dans la polycythémie vera. Il en résulte notamment une augmentation significative du taux d’hématocrite et donc du risque d’événements thromboemboliques.
La polycythémie vera (PV) est une néoplasie myéloproliférative chronique très rare, caractérisée par une hématopoïèse élevée. La majorité des patients atteints de PV présentent une mutation du gène de la tyrosine kinase JAK2 [1]. Il en résulte une augmentation de la prolifération cellulaire ainsi qu’une production accrue de cytokines pro-inflammatoires. La surproduction d’érythrocytes et l’augmentation de l’hématocrite qui en résulte augmentent la viscosité du sang. De cette manière, la survenue de thromboembolies est favorisée : 45% des décès liés à la PV sont dus à des complications thromboemboliques [2]. En principe, le pronostic est toutefois favorable. L’âge médian au moment du diagnostic est de 65 ans.
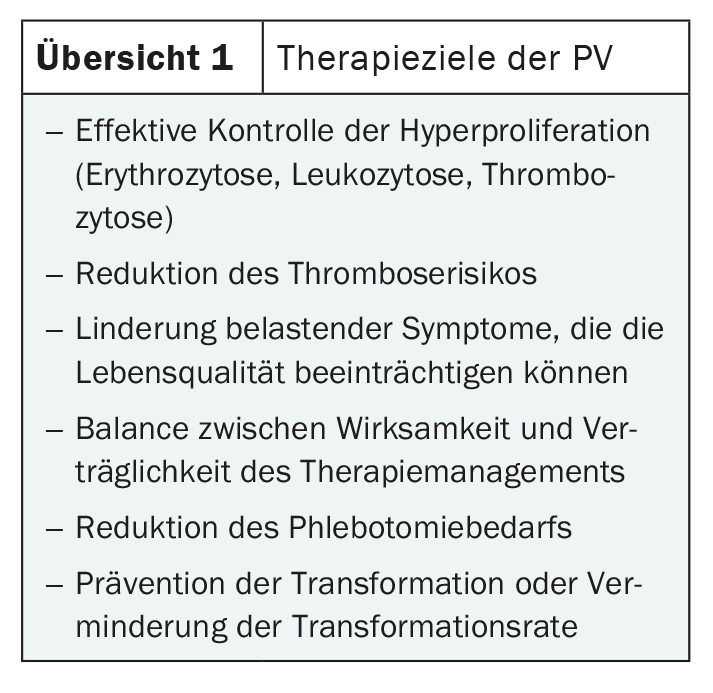
Bien que les symptômes soient variés, ils sont généralement assez peu spécifiques. C’est pourquoi le diagnostic n’est souvent posé que par hasard. Outre les maux de tête, les troubles de la vision, la fatigue et le prurit, les douleurs osseuses et les douleurs dans la partie supérieure de l’abdomen font partie des symptômes possibles. Celles-ci sont souvent causées par la splénomégalie typique de la PV [1,2]. L’augmentation de la masse des cellules sanguines peut entraîner des troubles de la circulation sanguine pouvant conduire à des thromboses veineuses et artérielles graves telles que l’embolie pulmonaire, l’apoplexie ou l’infarctus du myocarde. C’est pourquoi un diagnostic précoce et un traitement efficace sont indiqués (aperçu 1) [3].
Dans la phase chronique, qui dure généralement des années, les caractéristiques cliniques de l’augmentation de la myéloprolifération sont au centre des préoccupations. Les complications les plus fréquentes et potentiellement dangereuses sont les thromboembolies artérielles ou veineuses chez jusqu’à 40% des patients. Dans la phase tardive de la maladie, le principal problème réside dans une phase dite ‘spent’. Celle-ci se caractérise par une diminution de l’érythrocytose et une augmentation de la splénomégalie, associées à une fibrose de la moelle osseuse, qui peut être suivie d’une transformation en myélofibrose post-PV (secondaire) et/ou en leucémie aiguë [1]. Le taux global de MF post-PV est d’environ 15% après une période d’observation médiane de 10 ans et de 50% après 20 ans.
Traitement adapté au risque
La prévention des thromboembolies étant la priorité, la phlébotomie est souvent considérée comme le traitement de choix. Cela permet d’abaisser l’hématocrite (Hct) en dessous de 45% et de réduire l’hyperviscosité du sang. Des études ont montré que l’ajustement de l’Hct à moins de 45% peut réduire d’un quart le taux de décès cardiovasculaire lié à la PV [4]. Cependant, une phlébotomie est très fatigante. En outre, un traitement par acide acétylsalicylique (AAS) à faible dose doit être initialement mis en place. Ensuite, les recommandations thérapeutiques sont basées sur le score de risque. On peut considérer que le risque est faible chez les jeunes patients <60 ans qui n’ont pas eu de thrombose auparavant. La question de savoir si, dans certaines conditions, un traitement réducteur des cytokines devrait également être envisagé chez eux fait actuellement l’objet de discussions.

Cependant, la majorité des patients atteints de PV présentent de toute façon un risque élevé. Dans leur cas, l’introduction d’un traitement cytoréducteur est indiquée. L’hydroxyurée (HU) ou l’interféron alpha (INF) sont recommandés pour le traitement primaire [5]. Cependant, le HU en particulier ne convient pas à tous les patients et peut provoquer des effets secondaires graves (aperçu 2) [6]. C’est pourquoi l’INF est plus souvent utilisé chez les patients plus jeunes qui souhaitent avoir un enfant. Si le traitement de première ligne n’est pas toléré ou si les symptômes cliniques ne régressent pas suffisamment, le traitement doit être modifié. Des études ont montré que le ruxolitinib, un inhibiteur de JAK2, permettait de contrôler l’augmentation de la myéloprolifération tout en étant bien toléré [1]. De nombreux symptômes associés à la PV, tels que la fatigue et le prurit, ont également régressé. De plus, les effets ont été très rapides chez la plupart des patients, dans les quatre premières semaines. Le busulfan peut être utilisé comme traitement de substitution chez les patients d’âge avancé. Toutefois, le potentiel leucémogène de cette substance fait toujours l’objet de discussions, raison pour laquelle elle ne devrait être utilisée qu’avec modération. L’anagrélide peut être envisagé comme partenaire de combinaison avec, par exemple, HU ou INF. Celui-ci vise uniquement à réduire la production de plaquettes et peut agir en complément si les autres substances seules ne permettent pas d’obtenir des résultats satisfaisants.
Littérature :
- www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html (dernière consultation le 23.04.2024).
- Vannucchi AM, et al.: N Engl J Med 2015; 372: 426–435.
- Stein BL, Moliterno AR, Tiu RV: Polycythemia vera disease burden: contributing factors, impact on quality of life, andemerging treatment options. Ann Hematol 2014; 93: 1965–1976.
- Marchioli R, Finazzi G, Specchia G, et et al.: Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22–33.
- Barbui T, Tefferi A, Vannucchi AM, et al.: Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European Leukemia Net. Leukemia 2018; 32: 1057–1069.
- Barosi G, Birgegard G, Finazzi G, et al.: A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol 2010; 148: 961–963.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(2): 38









