Les cardiopathies congénitales sont les malformations d’organes les plus fréquentes chez les enfants. Toute malformation cardiaque nécessite une évaluation et une classification en cardiologie pédiatrique. Un suivi spécialisé en cardiologie pédiatrique est essentiel pendant toute l’enfance et l’adolescence, compte tenu de la croissance de l’enfant, des éventuels résultats cardiaques résiduels après l’intervention/l’opération et des éventuelles complications tardives.
Les cardiopathies congénitales sont les malformations d’organes les plus fréquentes, puisqu’elles touchent environ 1% des nouveau-nés. Pour la Suisse, cela correspondait en 2014 à environ 850 enfants pour 85 287 naissances vivantes. La gamme s’étend des malformations cardiaques simples, qui affectent peu le système cardiovasculaire, aux malformations graves (tableau 1) qui, si elles ne sont pas traitées, peuvent entraîner un décès prématuré [1].

La fréquence de survenue des différentes malformations cardiaques varie considérablement (tableau 2) et chaque malformation cardiaque nécessite un examen et une classification en cardiologie pédiatrique. La méthode d’examen diagnostique la plus importante est – outre l’examen clinique, y compris l’analyse de l’urine et des selles – l’analyse de l’urine et des selles. Auscultation – l’échocardiographie, qui permet de visualiser et de définir pratiquement toutes les structures cardiaques.

L’électrocardiogramme (ECG) (également appelé ECG Holter 24h/24) détecte les éventuels troubles du rythme ou les anomalies de conduction ainsi que les signes d’hypertrophie. Selon la situation, des méthodes d’examen spécifiques plus poussées telles que la spiroergométrie, l’imagerie par résonance magnétique cardiaque (IRMc), la tomodensitométrie (TDM) ou le cathétérisme cardiaque sont indiquées pour un diagnostic invasif (par exemple, mesure de la pression, angiographies). Cette dernière offre en même temps la possibilité d’une intervention. De nos jours, la plupart des malformations cardiaques sont ainsi diagnostiquées dès la première année de vie. En l’absence de traitement chirurgical ou interventionnel, les malformations cardiaques modérées et sévères réduisent considérablement l’espérance de vie.
Les progrès extraordinaires réalisés au cours des dernières décennies en chirurgie cardiaque, cardiologie interventionnelle, anesthésie et soins intensifs ont considérablement amélioré les chances de survie après une intervention cardiaque chez l’enfant. Ainsi, plus de 90% des enfants atteints de cardiopathie congénitale atteignent aujourd’hui l’âge adulte [2,3]. Cependant, ils sont souvent confrontés à de multiples problèmes au cours de leur vie en raison de leur physiopathologie cardiaque et des maladies qui les accompagnent souvent. Un point central est que la plupart des malformations cardiaques peuvent être réparées, mais pas corrigées à proprement parler [4].
Des résultats cardiaques résiduels importants et la croissance de l’enfant rendent essentiel un suivi spécialisé en cardiologie pédiatrique. Le suivi optimal des enfants atteints de maladies cardiaques nécessite une collaboration étroite entre les médecins généralistes et les pédiatres libéraux et les médecins spécialistes impliqués. Ceci est particulièrement important dans le cas de maladies associées complexes, qui affectent par exemple le développement neurologique [5].
A l’adolescence, la transition vers la médecine adulte vers un cardiologue spécifiquement formé (cardiologue GUCH ; GUCH = Grown-Ups with Congenital Heart Disease) doit être initiée à temps dans le sens d’un flux d’informations continu [6].
Complications au cours de l’évolution
Arythmies : Certaines anomalies cardiaques sont associées à des arythmies précoces. Par exemple, un bloc auriculo-ventriculaire complet peut se produire en cas de transposition cc des grandes artères. Mais le plus souvent, les troubles du rythme sont dus à des résidus hémodynamiques, tels qu’une charge de pression persistante avec fibrose ventriculaire ou charge de volume et/ou des cicatrices myocardiques (atrio-ventriculotomie), après une chirurgie cardiaque. Un trouble du rythme, y compris Trouble de la conduction, peut se développer tôt ou tard en postopératoire. On observe une tachycardie supraventriculaire (généralement une réentrytachycardie), un flutter ou une fibrillation auriculaire, une tachycardie auriculaire ectopique, un syndrome du sinus malade, une tachycardie ventriculaire, un bloc auriculo-ventriculaire et une mort subite.
Alors que le risque d’arythmie reste faible après des opérations simples sur le cœur, il augmente considérablement après des interventions complexes telles qu’une palliation de Fontan dans le cas d’un cœur univentriculaire. L’ECG (de longue durée) et l’ergométrie font donc partie de l’évaluation médicale régulière de ces patients. Pour les arythmies survenant tardivement après l’opération, il existe aujourd’hui, outre le traitement médicamenteux, l’électrophysiologie invasive qui donne de bons résultats grâce à l’ablation de cicatrices intracardiaques, par exemple.
Limitations physiques : Avec le nombre croissant d’enfants atteignant l’âge adulte après une réparation chirurgicale ou une palliation, l’inclusion des enfants dans le tissu social avec leurs pairs est devenue un objectif important. Alors qu’un patient en bonne santé cardiaque peut multiplier par cinq son débit cardiaque à l’effort, un patient souffrant d’une malformation cardiaque complexe, par exemple après une palliation de Fontan dans le cas d’un cœur univentriculaire, est au mieux capable de le doubler. Il en résulte une capacité physique limitée [7].
L’activité sportive dans le domaine de l’endurance est généralement encouragée. En revanche, les sports de force isométriques ou les sports compétitifs sont déconseillés chez la plupart des patients.
Le test et l’évaluation de la capacité physique par spiro-ergométrie font régulièrement partie du suivi cardiologique pédiatrique à partir de l’âge de dix ans environ.
Troubles de la croissance/développement neurocognitif : les enfants atteints d’une malformation cardiaque grave peuvent développer des troubles de la croissance et sont alors plus petits et plus maigres que les enfants sains du même âge. Diverses études ont montré que les enfants atteints de malformations cardiaques complexes présentent un risque accru de déficits neurocognitifs après une opération du cœur en période néonatale ou infantile. Un retard de développement de la motricité fine et globale, des troubles du comportement, une attention réduite, des symptômes d’hyperactivité et un retard de langage sont des déficits fréquents [6,8]. Il peut en résulter des effets négatifs sur la vie quotidienne et sur le parcours scolaire et professionnel à l’âge adulte. Les enfants atteints de malformations cardiaques légères à modérées ne sont généralement pas concernés. Il est important d’aborder ces points avec les parents et de proposer un soutien ciblé avec le pédiatre ou le médecin de famille qui les suit.
Endocardite : l’endocardite infectieuse est une complication grave et potentiellement mortelle chez les patients atteints d’une malformation cardiaque congénitale ou d’une cardiopathie acquise. C’est pourquoi l’éducation des parents et des patients lors du contrôle cardiologique pédiatrique est d’une grande importance. Les symptômes de l’endocardite doivent être reconnus à un stade précoce afin de pouvoir réagir de manière adéquate. De même, l’utilité et l’application correcte de la prophylaxie médicamenteuse de l’endocardite doivent être enseignées. Les directives actuelles pour la prophylaxie de l’endocardite chez les enfants en Suisse sont présentées dans le tableau 3.

Hypertension artérielle pulmonaire : toutes les cardiopathies congénitales avec de grands shunts intra- ou extracardiaques entraînent une charge de volume et de pression non entravée du lit vasculaire pulmonaire. En conséquence, une hypertension artérielle pulmonaire peut se développer, ce qui entraîne des changements irréversibles au stade tardif de cette maladie grave. Par exemple, dans le cas d’un grand défaut de septum ventriculaire non corrigé avec shunt gauche-droit, une augmentation de la pression dans le lit vasculaire pulmonaire se produit pendant des années, ce qui aboutit à une inversion du shunt avec cyanose due à un nouveau shunt droite-gauche (réaction d’Eisenmenger). L’objectif de l’intervention chirurgicale dans la petite enfance est, entre autres, d’empêcher ce développement. Les patients atteints d’une cardiopathie congénitale ont un risque accru de développer une hypertension artérielle pulmonaire, même en l’absence de shunts pertinents. La maladie a un impact négatif sur la qualité et la durée de vie, en fonction de sa gravité [10]. Pour le diagnostic et la mise en place d’un traitement spécifique, il est généralement nécessaire de s’adresser à un centre spécialisé.
Les maladies associées : Les cardiopathies congénitales sont associées dans 15-20% des cas à d’autres malformations et/ou anomalies chromosomiques. Les enfants atteints de trisomie 21 présentent un vitium cordis dans 40 à 50 % des cas. Ces patients, qui présentent souvent des malformations complexes, bénéficient d’une prise en charge multidisciplinaire.
Exemples de suivi et d’aspects spécifiques de certaines malformations cardiaques
Coarctation de l’isthme aortique (AIST) : l’objectif est d’éliminer précocement la sténose et de créer une aorte aussi libre de gradient et de calibre normal que possible. Cela ne peut être réalisé que par chirurgie ou par cathétérisme cardiaque interventionnel (dilatation par ballonnet ou implantation d’endoprothèse) (Fig. 1). La méthode de traitement dépend de l’âge du patient ainsi que de la morphologie de l’AIST. Il existe souvent des malformations associées (par exemple, une valve aortique bicuspide dans environ 50% des cas).
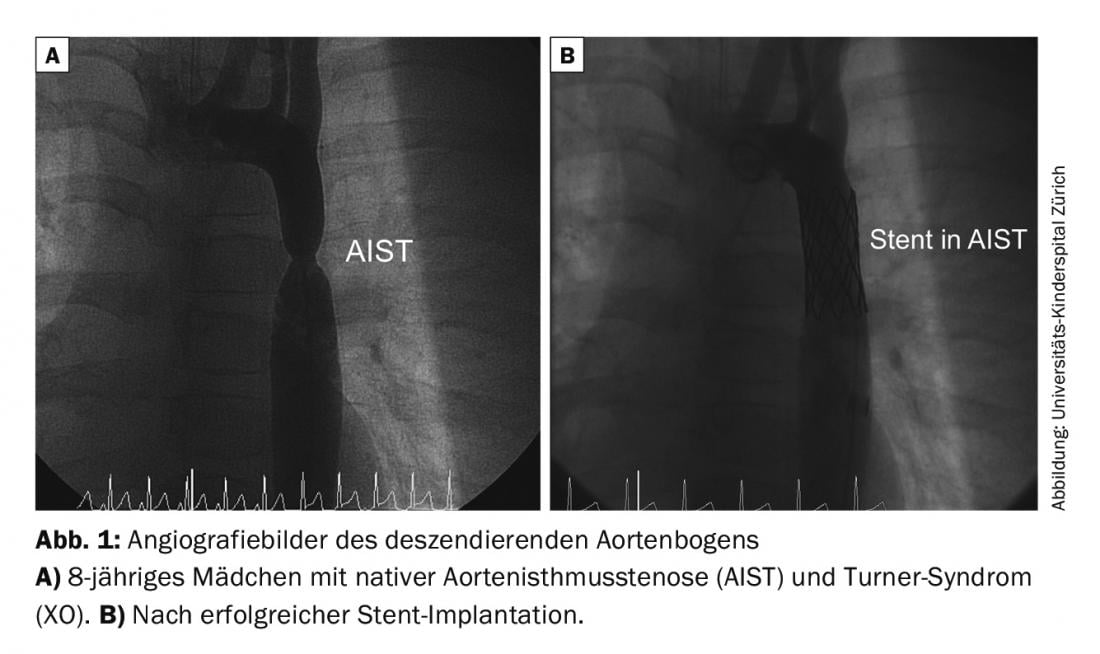
Les patients ayant subi une réparation chirurgicale ont un risque de 5-10% de re-sténose au cours de l’évolution. Le risque d’hypertension artérielle augmente avec l’âge, même si la sténose a été efficacement éliminée. D’autres risques possibles sont la formation d’anévrismes/dissections aortiques au niveau de la suture ou du stent.
Recommandations pour le suivi [11]:
- en général annuellement, au moins une fois par an tous les 2 ans :
- statut clinique
- Mesure de la pression artérielle (PA) sur les 4 membres
- selon la BD au repos : mesure de la BD sur 24h
- ECG
- Echocardiographie (taille du ventricule gauche, épaisseur et fonction du myocarde, degré de sténose sur l’arc aortique/l’isthme, attention aux malformations associées le cas échéant)
- Ergométrie (à partir de 10 ans) tous les 3-4 ans
- IRMc : en cas de visualisation insuffisante à l’échocardiographie : visualisation de l’arc aortique, exclusion de la formation d’anévrisme tous les 5 ans
d-transposition des grosses artères (d-TGA) : De nos jours, l’opération de switch artériel visant à rétablir la concordance ventriculo-artérielle de la d-TGA est la méthode de traitement de premier choix. L’aorte et l’artère pulmonaire sont interverties et, grâce à la manœuvre dite de Lecompte, la bifurcation pulmonaire se trouve finalement devant l’aorte (Fig. 2). Parallèlement, une réimplantation des artères coronaires est nécessaire. La valve pulmonaire anatomique sert donc à vie de valve néo-aortique dans la circulation systémique.
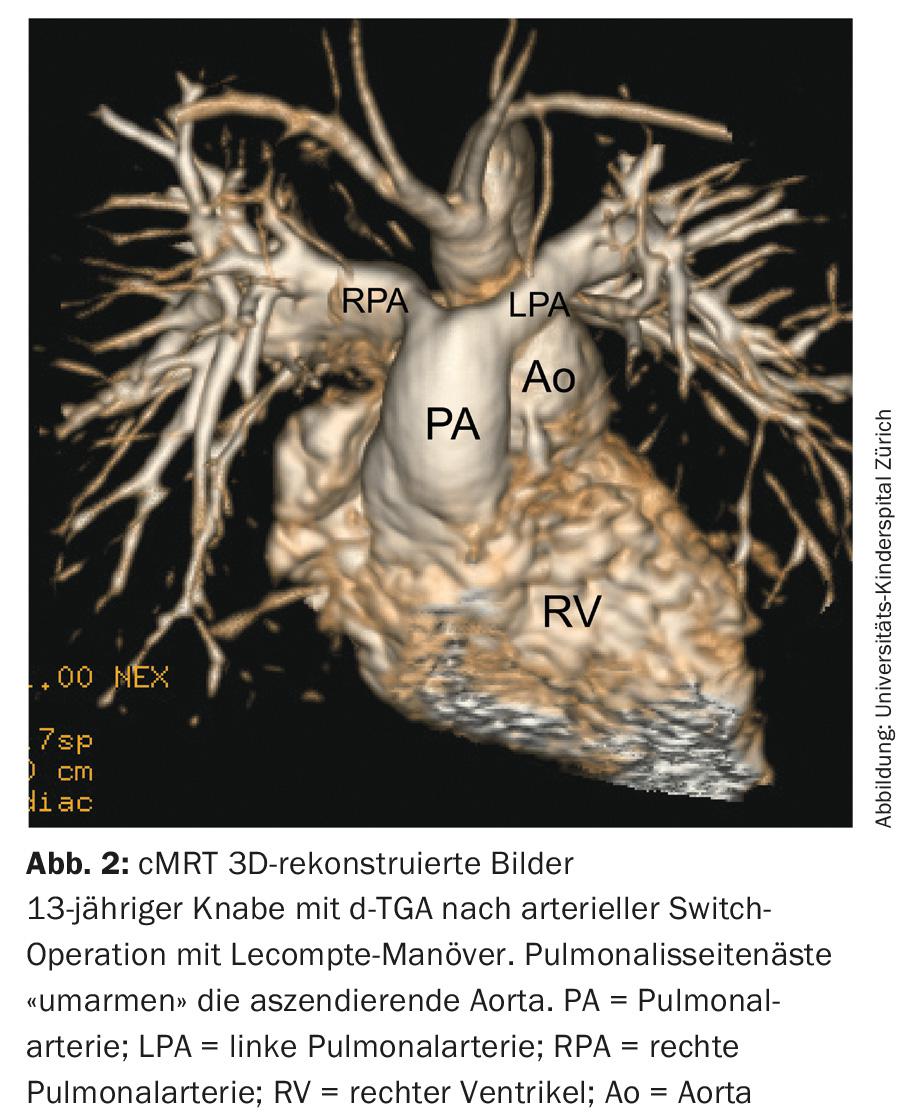
À long terme, cette néo-valve aortique peut devenir dysfonctionnelle et la racine de l’aorte peut se dilater. En raison de la manœuvre de Lecompte avec déplacement des branches latérales pulmonaires antérieures à l’aorte ascendante, une torsion et un étirement peuvent se produire, entraînant une sténose des branches latérales pulmonaires. Comme l’opération implique un transfert coronaire, la torsion et l’étirement peuvent également entraîner une sténose ou, dans les cas extrêmes, une occlusion d’une artère coronaire, avec infarctus du myocarde et mort subite cardiaque consécutifs.
Recommandations pour le suivi [9,11]:
- en général annuellement, au moins une fois par an tous les 2 ans :
- statut clinique
- ECG
- Échocardiographie (taille et fonction du ventricule, degré de sténose des branches de la néo-lisière pulmonaire, trouble de la mobilité de la paroi, fonction de la valve néo-aortique, dimension de la racine néo-aortique)
- ECG à long terme selon l’évaluation individuelle
- Coronarographie en cas d’évolution périopératoire compliquée, de patient symptomatique ou de diagnostic de routine anormal.
- Ergométrie (à partir de 10 ans) tous les 2-3 ans
- IRMc : en cas de mauvaise qualité sonore, pour évaluer la taille et la fonction du ventricule droit et du ventricule gauche, ainsi que pour évaluer les branches de la dissection pulmonaire. Évaluation des artères coronaires et/ou de la cicatrisation myocardique.
Tétralogie de Fallot (TOF) : l’objectif de la réparation chirurgicale anatomique de la tétralogie de Fallot est la fermeture par patch de la CIV et la suppression de l’obstruction de la voie de sortie du ventricule droit (RVOTO). En cas de petit diamètre de l’anneau de la valve pulmonaire et/ou de valve dysplasique, l’implantation d’un patch transanulaire est nécessaire, ce qui entraîne toujours une insuffisance valvulaire pulmonaire (Fig. 3). Celle-ci entraîne à moyen et long terme une dilatation importante du ventricule droit, qui peut conduire à un dysfonctionnement ventriculaire, à une diminution des performances et à une arythmie, et finalement à la nécessité d’un remplacement de la valve pulmonaire.

Le risque de bloc auriculo-ventriculaire complet nécessitant l’implantation d’un pacemaker est faible (<2%).
Recommandations pour le suivi [9,11]:
- en général annuellement
- statut clinique
- ECG (largeur QRS)
- Echocardiographie (RVOTO résiduelle, y compris les branches de la dissection pulmonaire, insuffisance valvulaire pulmonaire, taille et fonction du ventricule droit, DSV résiduel, taille et fonction du ventricule gauche, dimension de la racine aortique)
- ECG à long terme (tachycardie non soutenue) min. tous les 3 ans chez les patients asymptomatiques
- Ergométrie (à partir de 10 ans), tous les 3-5 ans
- IRMc : en cas de dilatation croissante du VR : pour évaluer la taille et la fonction du ventricule droit ainsi que pour évaluer l’insuffisance pulmonaire (fraction de régurgitation), la taille et la fonction du ventricule gauche. Évaluation des branches de la dissection pulmonaire.
Single Ventricle (SV) : parmi les cœurs univentriculaires, on distingue fondamentalement deux formes de base, selon que le ventricule droit ou le ventricule gauche ne s’est pas suffisamment développé (p. ex. atrésie tricuspide ou syndrome du cœur gauche hypoplasique) ; Fig. 4). Le traitement chirurgical sous forme de trois interventions palliatives échelonnées en général se termine par la “circulation de Fontan”. L’objectif est de créer un circuit en série à partir des circuits parallèles existants jusqu’à présent. En période néonatale, il faut d’abord stabiliser la circulation parallèle, ce qui implique généralement, selon la malformation cardiaque, la mise en place d’un shunt ou la réduction de l’hyperperfusion pulmonaire par ligature. Dans un deuxième temps, une anastomose cavopulmonaire supérieure (de Glenn) bidirectionnelle est créée chez le nourrisson, ce qui entraîne une nette réduction du volume du cœur. Avec la complétion de Fontan à l’âge de 2-3 ans au moyen d’un conduit extracardiaque, l’anastomose cavopulmonaire totale, la palliation définitive est atteinte chez les patients à cœur univentriculaire. Ce n’est qu’après cette séparation définitive de la circulation que la cyanose est supprimée (à l’exception de petits shunts résiduels ; collatérales ou fenestration).
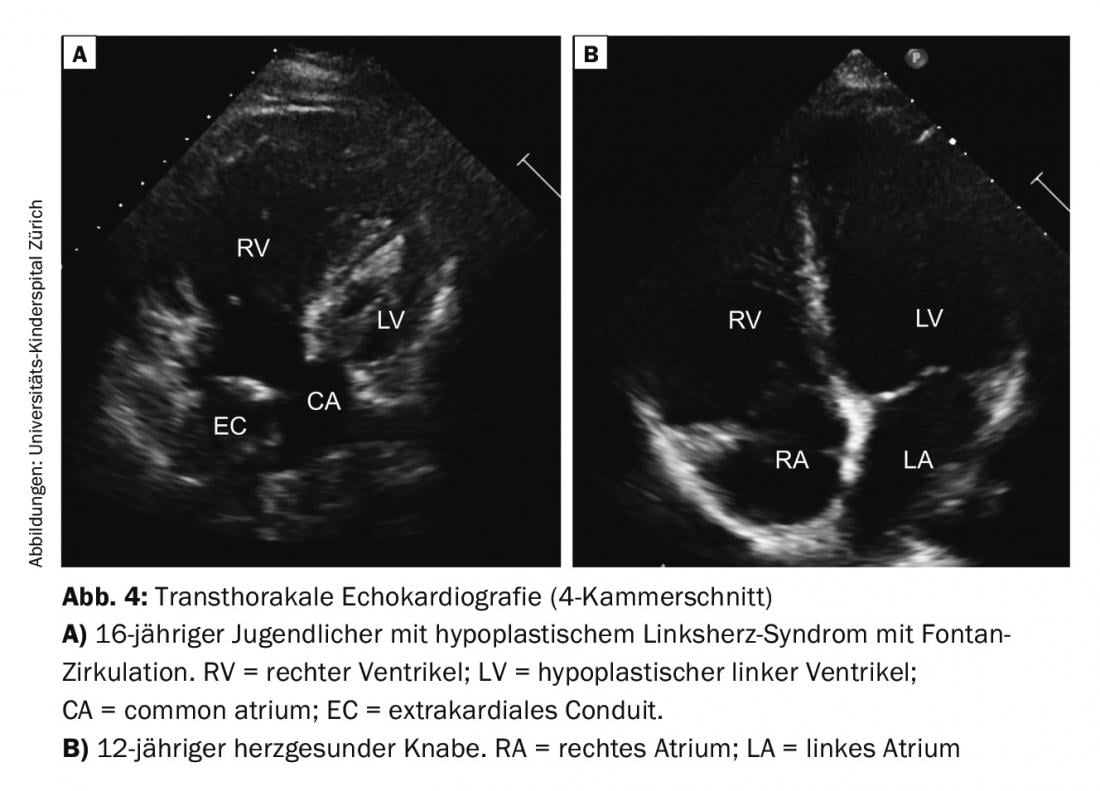
La prise en charge pendant les différentes étapes de l’opération et le suivi de ce groupe de patients hétérogène et unique, avec toutes ses complications et ses conséquences tardives, constitue un défi majeur pour les cardiologues pédiatriques et, plus tard, pour les cardiologues spécialisés pour adultes (cardiologue GUCH).
Les complications chez ces patients sont très variées : fonction ventriculaire réduite, insuffisance valvulaire AV, sténose dans la circulation systémique ou pulmonaire, arythmie, incompétence chronotrope, thrombose/embolie, entéropathie par perte de protéines, bronchite plastique, dysfonction hépatique, hypertension pulmonaire, parésie diaphragmatique, scoliose, etc.
Recommandations pour le suivi après l’achèvement d’une fontan [9,11]:
- min. annuel
- statut clinique
- Mesure de la BD sur les 4 membres
- Saturation en oxygène transcutanée
- ECG
- Echocardiographie (fonction SV, insuffisance valvulaire AV, obstruction gauche (sténose sous-aortique, sténose isthmique), profil de flux de l’anastomose cavopulmonaire supérieure ou dans le circuit du fontan et des veines hépatiques, etc.)
- Examen de laboratoire : protéines totales ou albumine sérique, bilan hépatique, pro-BNP comme paramètre de la fonction cardiaque, hémoglobine.
- ECG à long terme (réentrée intra-auriculaire non soutenue, compétence chronotrope), en cas d’indices de troubles du rythme ou au moins tous
- 3 ans
- Ergométrie (à partir de 10 ans), min. tous les 3 ans
- IRMc : évaluation de la fonction ventriculaire, quantification du flux sanguin, représentation anatomique des connexions de Fontan. Réalisation en fonction de l’évolution clinique, mais généralement recommandée à l’adolescence et avant la transition vers les cardiologues du GUCH.
- Cathétérisme cardiaque diagnostique : en cas de détérioration de la circulation de la fontanelle, comme par exemple une diminution de la capacité d’effort, une cyanose croissante, une ascite ou une entéropathie par perte d’albumine, une bronchite plastique, etc.
Mot de la fin
De nos jours, la plupart des cardiopathies congénitales ont un bon pronostic à long terme. La détection précoce de la malformation cardiaque, le traitement chirurgical et/ou interventionnel par une équipe expérimentée ainsi que le suivi attentif y contribuent de manière décisive. Une étroite collaboration entre le pédiatre et le cardiologue pédiatrique, ainsi que d’autres spécialistes si nécessaire, et plus tard la transition vers un cardiologue GUCH, est essentielle.
Littérature :
- Hoffmann JIE, et al : L’incidence des cardiopathies congénitales. JACC 2002 ; 39(12) : 1890-1900.
- Warnes CA, et al : Task Force 1 : The Changing Profile of Congenital Heart Disease in Adult Life. JACC 2001 ; 37(5) : 1161-1198.
- Moons P, et al : Temporal Trends in Survival to Adulthood Among Patients Born with Congenital Heart Disease from 1970 to 1991 in Belgium. Circulation 2010 ; 122 : 2264-2272.
- Oechslin E : Malformations cardiaques congénitales : soins spécialisés à vie pour une maladie chronique. Médecine cardiovasculaire 2006 ; 9 : 373-375.
- Mackie A : Enfants et adultes atteints d’une cardiopathie congénitale perdus pour le suivi. Circulation 2009 ; 120(4) : 302-309.
- Bauersfeld U : Transition, transfert et coopération chez les patients atteints de cardiopathies congénitales – collaboration continue entre la cardiologie pédiatrique et la cardiologie adulte. Médecine cardiovasculaire 2006 ; 9 : 336-341.
- Gewillig M, et al. : Échec de la circulation de Fontan. Heart Failure Clin 2014 ; 10(1) : 105-116.
- Sterken C, et al : Développement neurocognitif après une chirurgie cardiaque pédiatrique. Pediatrics 2016 ; 137(6), doi : 10.1542/peds.2015-4675.
- Wernovsky G, et al : Guidelines for the Outpatient Management of Complex Congenital Heart Disease. Congenit Heart Dis 2006 ; 1 : 10-26.
- Dimopoulos K, et al : Hypertension pulmonaire liée à une cardiopathie congénitale : un appel à l’action. Eur Heart J 2014 ; 35(11) : 691-700.
- Société allemande de cardiologie pédiatrique, éd. Schmaltz AA : Lignes directrices pour le diagnostic et le traitement en cardiologie pédiatrique. Elsevier GmbH, Urban & Fischer Verlag 2007.
- Knirsch W, et al. : Nouvelles recommandations pour la prophylaxie antibiotique de l’endocardite chez les enfants en Suisse. Paediatrica 2009 ; 20(4) : 28-34.
CARDIOVASC 2016 ; 15(5) : 4-9