
Une éosinophilie sanguine est définie par un nombre >500 éosinophiles par μl. Si un nombre >1500/μl est mesuré au moins deux fois en deux semaines, on a affaire à une hyperéosinophilie (HE). Il s’agit d’un syndrome hyperéosinophilique (SHE) lorsqu’une lésion d’organe s’ajoute à l’HE. La manière de classifier davantage les HES et de diagnostiquer par exemple une EGPA a été discutée lors du congrès DGIM 2023.
Depuis 2012, les HE et HES sont divisés en formes primaires et réactives sur la base de différentes étiologies, ainsi qu’en HE et HES idiopathiques pour lesquelles aucune maladie clonale ou réactive sous-jacente n’est identifiée. En raison du nombre croissant de marqueurs et de cibles, une mise à jour de cette classification a été publiée au début de l’année [1]. Dans la mise à jour, il a notamment été précisé que pour définir une HE, il faut un nombre >1500/µl mesuré à au moins deux reprises à un intervalle d’au moins deux semaines – dans la version précédente, l’intervalle était de quatre semaines.
Comme l’écrivent le professeur Peter Valent, de l’hôpital universitaire AKH de Vienne, et ses collègues, en plus de la présence d’HE dans le sang et/ou les tissus et des lésions organiques associées à l’HE comme troisième critère, l’HES est défini par l’exclusion de toute autre maladie ou pathologie sous-jacente comme cause primaire des lésions organiques.
Les auteurs soulignent que l’HES n’est ni un diagnostic définitif ni une maladie immunologique ou hématologique définie. Au contraire, l’étiologie contributive doit être identifiée, si possible, chez tous les patients atteints de SHE. Si aucune maladie sous-jacente n’est identifiée, le diagnostic final est l’HES idiopathique (HESI).
Les sous-types de l’HES sont les maladies myéloprolifératives (14%) et lymphoprolifératives (14%), les maladies idiopathiques (42%), les maladies associées (9%) et les maladies superposées (21%). Le groupe des maladies associées comprend notamment la granulomatose éosinophile avec polyangéite (EGPA), autrefois connue sous le nom de syndrome de Churg-Strauss.
Des anticorps cytoplasmiques anti-neutrophiles (ANCA) sont détectés chez environ 40% des patients atteints d’EGPA. Compte tenu de la similitude des manifestations cliniques de l’EGPA (et d’autres syndromes liés à l’HE) avec les caractéristiques classiques de l’HES, Valent et al. a conclu que ces syndromes devraient être classés comme HES si les critères HES sont remplis. Si ce n’est pas le cas, le diagnostic final devrait être le syndrome correspondant et non HES.
| Critères de classification ACR pour la vascularite Eosinophilie >10% (facultatif) Éosinophilie tissulaire Asthme (>90%) Anomalies des NNH (environ 50%) Infiltrats pulmonaires fugaces (50-70%) Neuropathie (environ 75%) |
| d’après [3] |
La détection de la vascularite est souvent impossible
“Pour faire la différence entre un HES et une EGPA, il faut savoir que l’EGPA nécessite la preuve de la vascularite”, a expliqué le Dr Christof Iking-Konert, chef du service de rhumatologie à l’hôpital municipal Triemli de Zurich [2]. Il a fait référence à cette question. Sur les critères de classification de l’American College of Rheumatology (ACR) [3] (encadré).
L’EGPA est la forme la plus rare de vascularite associée aux ANCA, avec une prévalence de 24/1 million. Le premier défi diagnostique est donc de commencer par y penser. Le deuxième obstacle : La détection nécessaire d’une vascularite à l’histologie est souvent difficile, par exemple parce qu’une biopsie n’est pas réalisable chez des patients très malades. La plupart du temps, l’EGPA n’est que suspectée, c’est pourquoi on s’oriente vers des substituts de vascularite, comme l’a expliqué le Dr Iking-Konert. Selon lui, si un ou plusieurs de ces facteurs sont présents, on peut supposer qu’il s’agit d’un corrélat de vascularite :
- Hémorragie alvéolaire
- Purpura palpable
- Infarctus du myocarde suite à une coronarite avérée
- Hématurie associée à des cylindres d’érythrocytes ou >10% d’érythrocytes dysmorphiques ou hématurie et 2+ protéinurie
- Mononévrite ou mononévrite multiplex
- MPO ANCA et tout type de manifestation systémique.
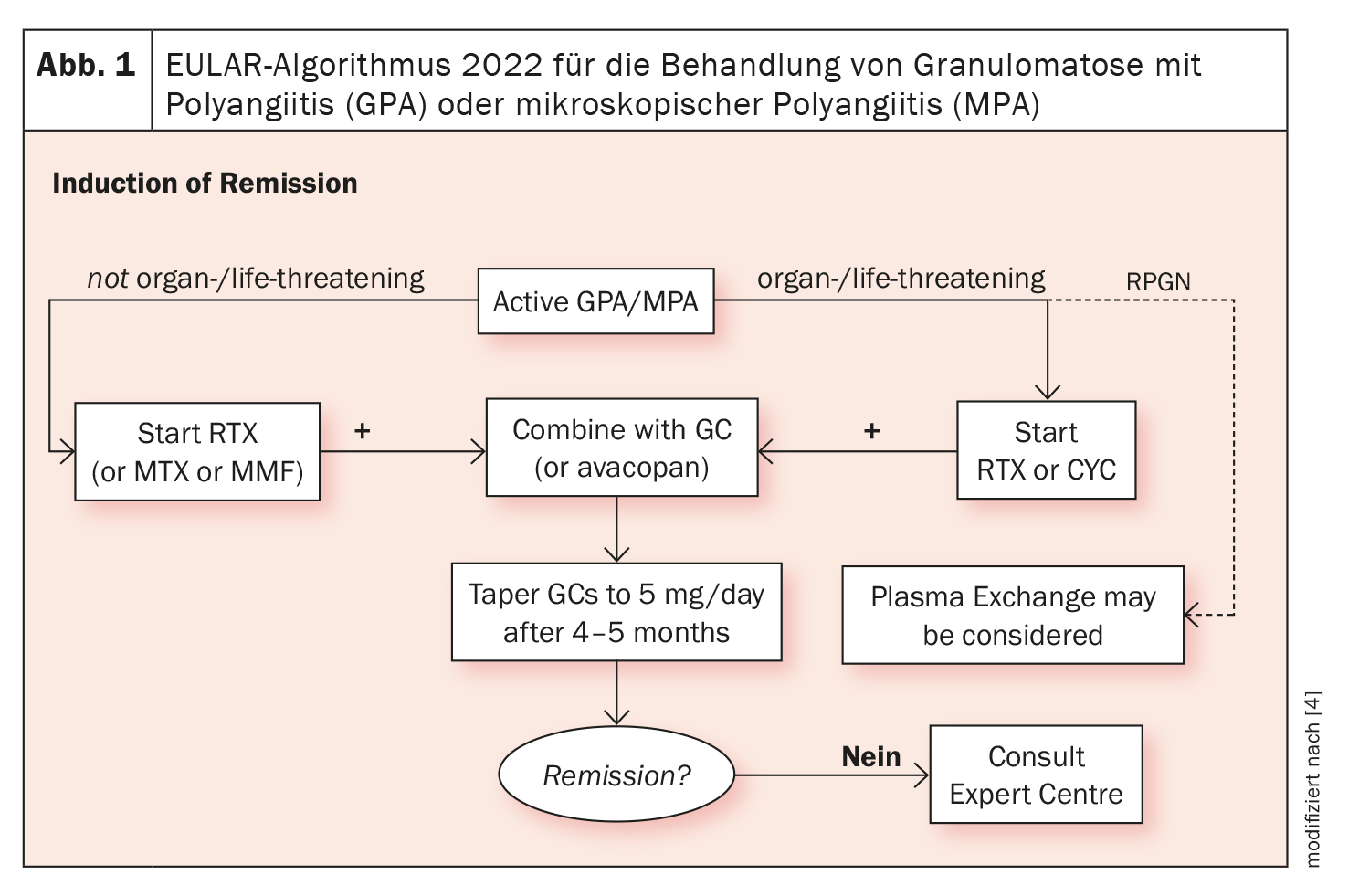
Des médicaments biologiques sont disponibles depuis plus d’une décennie pour le traitement de l’EGPA. Outre le rituximab, les thérapies ciblées avec des anticorps IL-5 se sont établies. Le Dr Iking-Konert a fait référence à la mise à jour en 2022 du guide EULAR sur la prise en charge des vascularites associées aux ANCA : Il y est recommandé de commencer un traitement par cyclophosphamide (CYC) ou rituximab (RTX) associé à de la cortisone en cas d’EGPA mettant en danger les organes ou le pronostic vital. Dans les maladies ne mettant pas en jeu le pronostic vital ou celui d’un organe, le blocage de l’IL-5 est indiqué en cas d’évolution réfractaire ou sévère (figure 1) [4].
Congrès : DGIM Industriesymposium GSK
Littérature :
- Valent P, et al.: Allergy 2023; 78: 47–59.
- Industrie Symposium «Und täglich grüsst das Immunsystem: seltene Autoimmunerkrankungen erkennen und behandeln». 129. Kongress der DGIM, 23.04.2023; Veranstalter: GSK.
- Jennette JC, et al.: Arthritis Rheum 1994; 37: 187–192.
- Hellmich B, et al.: Ann Rheum Dis 2023; doi: 10.1136/ard-2022-223764.
| Image de couverture : Micrographe à haute magnitude de la vascularite éosinophilique associée au syndrome de Churg-Strauss, abrégé CSS. H&E stain. Source : Nephron, wikimedia |
InFo RHEUMATOLOGIE 2023; 5(1): 17









