Un article de synthèse qui se concentre spécifiquement sur les manifestations cutanées possibles au niveau des mains et des pieds dans les collagénoses classiques, telles que le lupus érythémateux, la sclérodermie systémique, la dermatomyosite et le syndrome de Sharp.
Il est bien connu que les collagénoses classiques que sont le lupus érythémateux, la sclérodermie systémique, la dermatomyosite et le syndrome de Sharp (synonyme : collagénose mixte) peuvent provoquer divers symptômes cutanés et lésions organiques. Cet article de synthèse se concentre exclusivement et spécifiquement sur la zone des mains et des pieds, respectivement sur les manifestations cutanées possibles sur les mains et les pieds en cas de collagénose.
Phénomène de Raynaud
Le phénomène de Raynaud (synonyme : maladie de Raynaud, syndrome de Raynaud) est commun à presque toutes les collagénoses, c’est pourquoi il doit être placé avant les autres descriptions en tant que manifestation cutanée supérieure sur les mains et les pieds. Il s’agit typiquement d’un jeu caractéristique de trois couleurs des mains : blanc, bleu, rouge, déclenché par le froid ou le stress émotionnel, d’où le nom de phénomène tricolore . Elle est due à une mauvaise régulation sympathique du système nerveux autonome qui, par l’intermédiaire de récepteurs, provoque une vasoconstriction excessive des artérioles, ce qui entraîne d’abord une ischémie avec insensibilité et douleur dans les zones concernées (blanc), puis une cyanose due à l’hypoxie (bleu). La plupart du temps, ces spasmes se résolvent d’eux-mêmes, entraînant une hyperémie réactionnelle finale avec rougeurs et picotements (rouge). Seule cette succession de couleurs caractéristiques, ainsi décrite, est définie comme un phénomène de Raynaud et doit être décrite de la même manière dans la documentation de l’anamnèse. Le syndrome de Raynaud primaire doit être distingué du syndrome de Raynaud secondaire. Le syndrome de Raynaud primaire, beaucoup plus fréquent, ne présente pas de maladie sous-jacente causale ou concomitante, contrairement au syndrome de Raynaud secondaire, qui peut être distingué par son aspect clinique, avec une atteinte asymétrique de certains doigts et la détection d’anticorps antinucléaires. L’apparition du phénomène de Raynaud dans les collagénoses est donc, par définition, la forme secondaire.
Sclérodermie systémique
La sclérodermie systémique (sclérose) se caractérise par l’apparition précoce de lésions cutanées caractéristiques au niveau des mains et des pieds, ce qui permet souvent de suspecter une sclérodermie systémique.
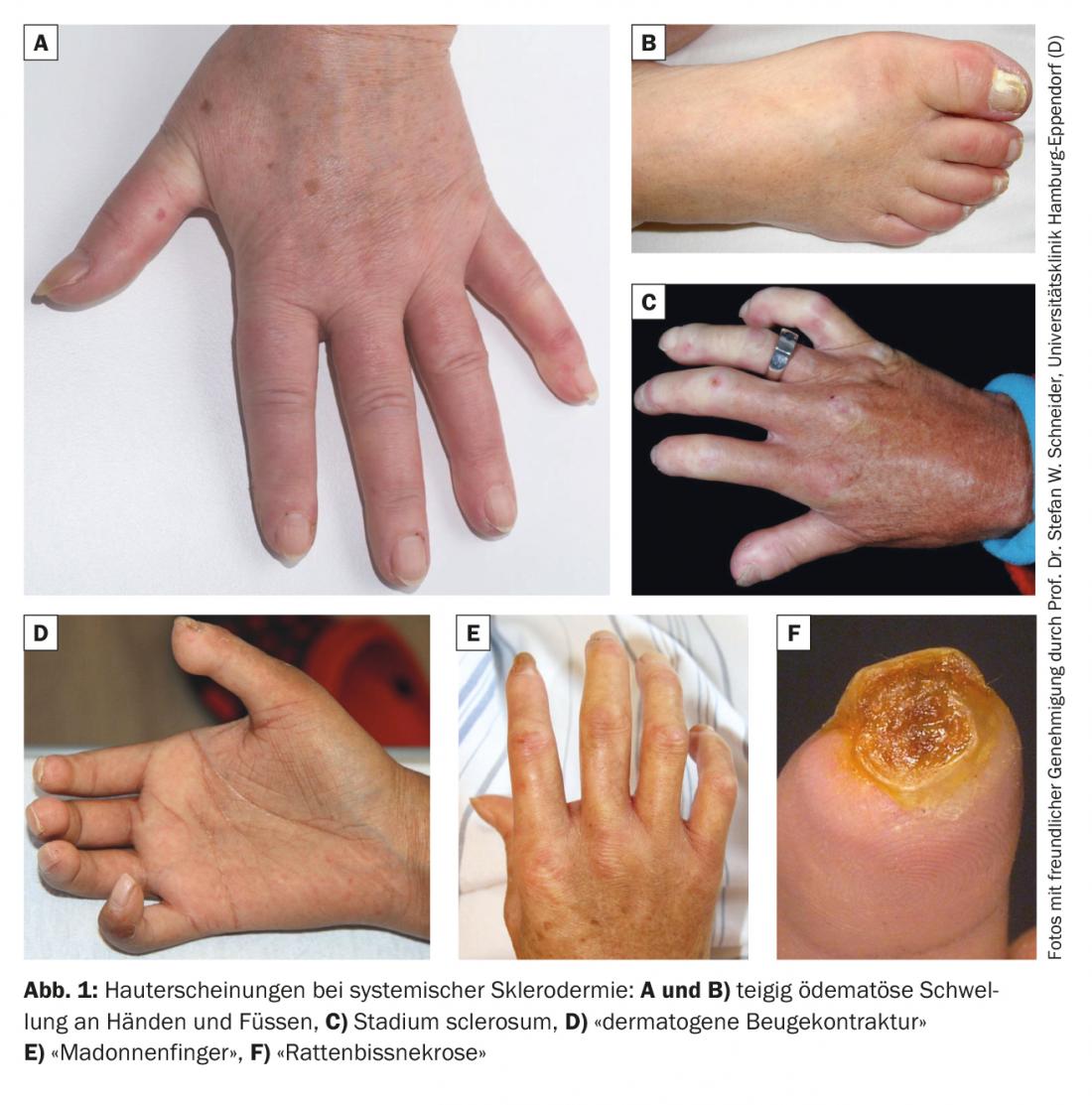
On distingue cliniquement un stade oedematosum avec des doigts gonflés de pâte et d’œdème (Fig. 1A et 1B) du stade sclérosé, qui se développe plus tard, avec une peau des doigts cireuse et non mobile (sclérodactylie). (Figure 1C). Au stade final, les doigts se fixent et se tiennent comme des griffes (“contractures de flexion dermatogènes”). (Fig. 1D). Les doigts deviennent également beaucoup plus fins vers l’extrémité des doigts, ce qui est bien décrit par le terme “doigts de madone”. (Figure 1E). L’extrémité distale peut présenter des troubles trophiques et des nécroses (“nécroses par morsure de rat”). (Fig. 1F). Les ongles peuvent également présenter des changements trophiques correspondants.
En microscopie capillaire, toutes les pathologies peuvent être observées chez les patients atteints de sclérodermie, parfois même simultanément. Cependant, on peut typiquement distinguer différents stades (aperçu 1).

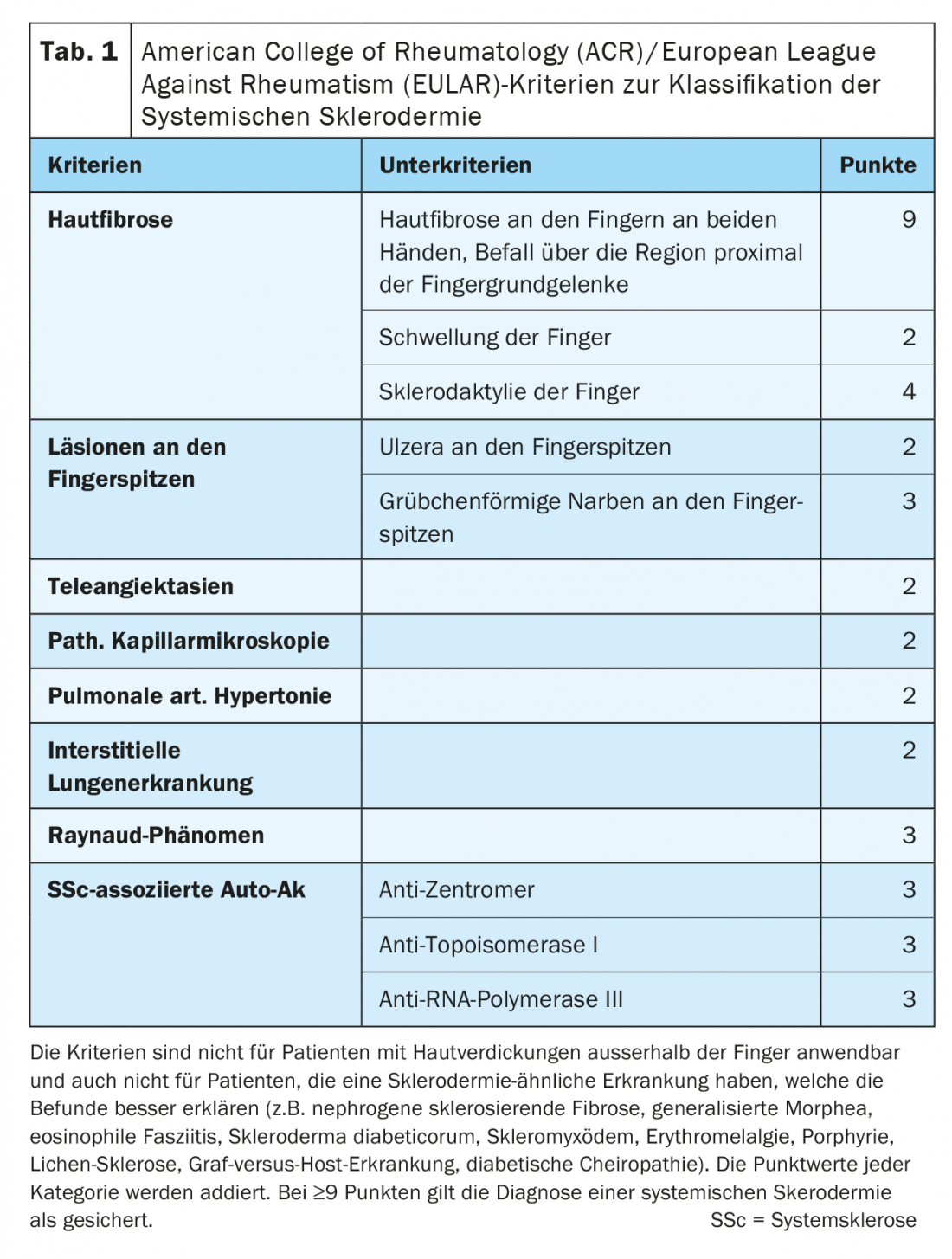
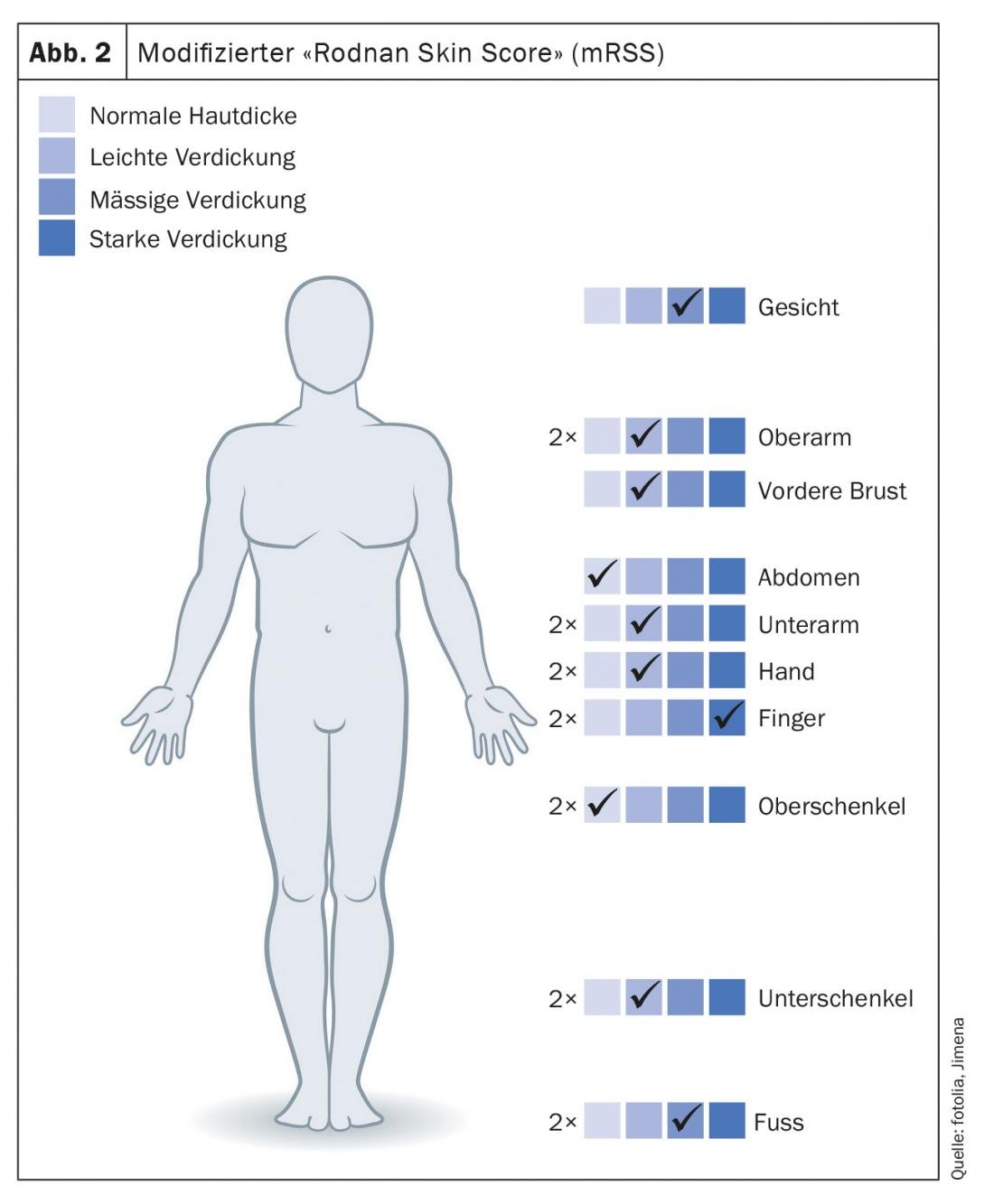
Les modifications de la peau des mains et des pieds, si typiques de la sclérodermie systémique, sont également prises en compte dans les “scores” et les classifications pour le diagnostic et la classification de la gravité. Sur la base des critères de Collège américain de rhumatologie (ACR)/Ligue européenne contre le rhumatisme (EULAR), la sclérodermie systémique peut être diagnostiquée presque uniquement sur la base des modifications de la peau des mains. (tableau 1). Le “score cutané de Rodnan” modifié (mRSS) de 1995 est toujours considéré comme le paramètre de corrélation le mieux validé pour estimer l’implication des organes, en particulier les poumons. Dans 17 régions, les mains et les pieds étant représentés six fois au total, la sclérose cutanée est évaluée par un score de 0 à 3 (figure 2).
Lupus érythémateux disséminé (LES)
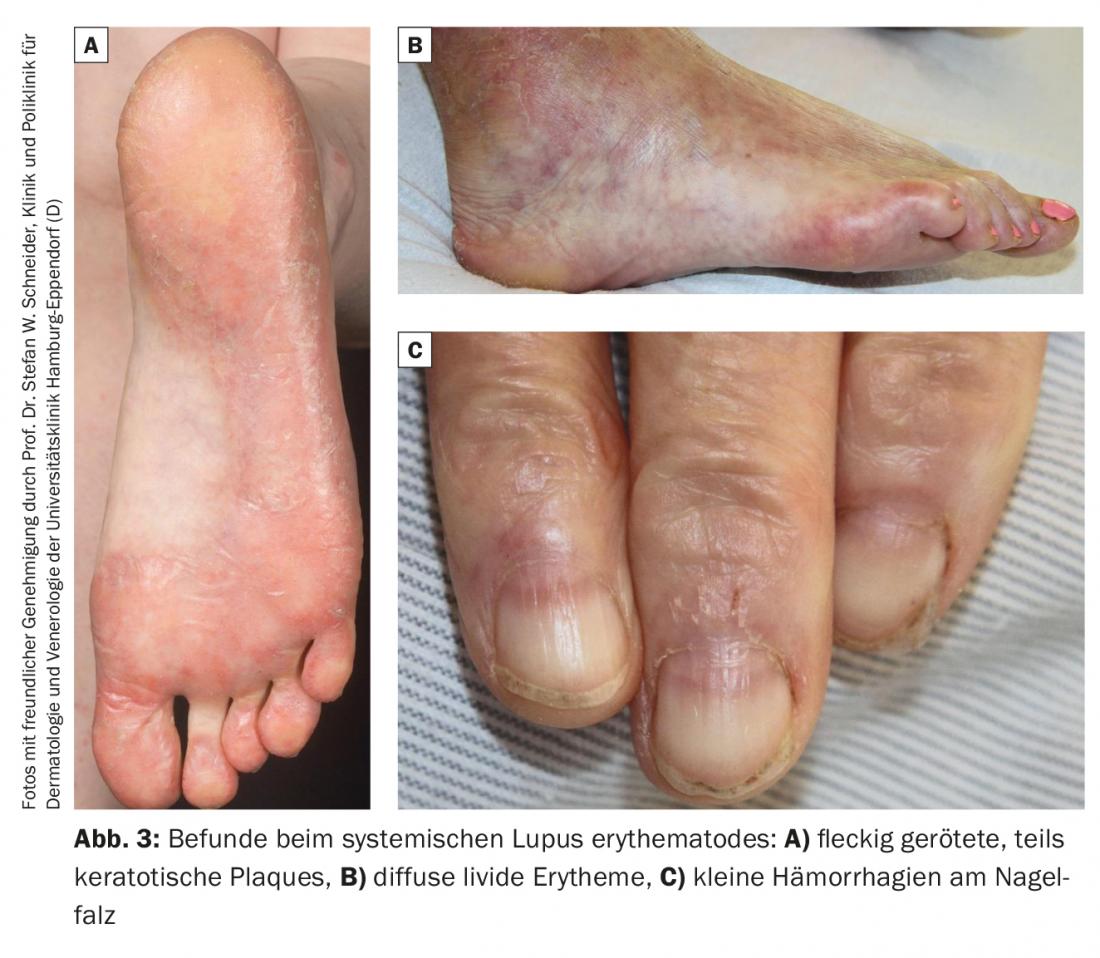
Les lésions cutanées des mains et des pieds sont moins spécifiques dans le lupus érythémateux disséminé que dans la sclérodermie systémique. On observe des livedo racemosa, des plaques rouges tachetées, parfois kératosiques, sur la face dorsale des doigts ou sur les plantaires (figure 3A), des érythèmes livide diffus sur les palmes et les plantaires (figure 3B) ainsi que sur les phalanges et les orteils, des télangiectasies périunguéales et au bout des doigts ainsi que de petites hémorragies sur le repli de l’ongle (figure 3C).
En microscopie capillaire, les patients atteints de LED présentent généralement une image colorée avec une accumulation de différentes modifications pathologiques. Ainsi, on trouve principalement des variations de calibre, des “sludge”, des ramifications et des élongations.
Chilblain-Lupus
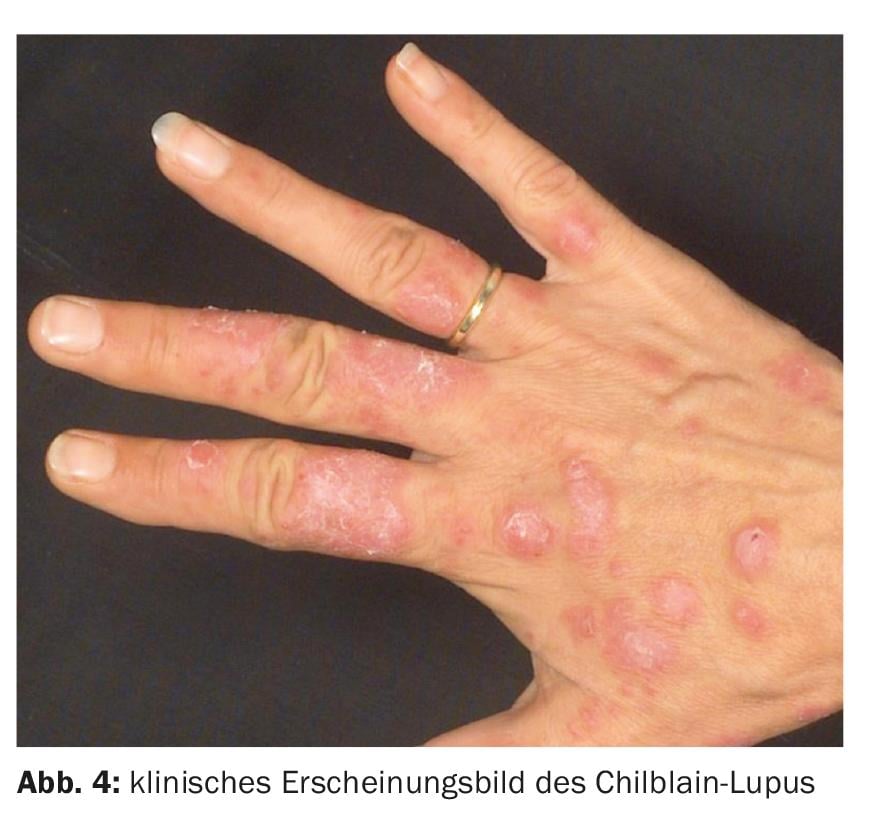
Le lupus de Chilblain est une variante rare et acquise du lupus érythémateux cutané, qui se caractérise par des plaques dolentes à la pression, induites par le froid et localisées au niveau de l’aréole, ce qui explique qu’il passe souvent inaperçu et n’est pas diagnostiqué. Il survient presque exclusivement chez les femmes, surtout pendant la saison froide et sous des latitudes plus froides, avant, avec ou après d’autres manifestations de lupus érythémateux cutané ou systémique.
Au stade précoce, on observe des enflures, des plaques et des nodules dolents à la pression, d’un rouge profond, qui rappellent les engelures (chilblain = engelure).
Au fil des années, des plaques hyperkératosiques rougeâtres et livide, aux limites floues et nettement indurées, se développent, soit lisses en surface, soit squameuses à grosses lamelles. Des hyperesthésies, des atrophies centrales, des érosions ou des rhagades sur les parties dorsales des articulations des doigts ou des ulcérations étendues peuvent apparaître (figure 4). L’évolution saisonnière est typique, avec une nette accentuation des symptômes cutanés pendant la saison froide et une rémission tout aussi nette, mais généralement incomplète, pendant les mois d’été.
En 2006, une forme familiale de lupus de Chilblain, due à une mutation du gène TREX1, a été décrite pour la première fois.
Le laboratoire révèle le plus souvent une hypergammaglobulinémie. ANA : souvent positif ; Ac antiphospholipides, anti SSA/Ro variable positif ; ADNdb : négatif ; BSR accéléré. Variable : Diminution de C3 et C4. Le diagnostic est considéré comme confirmé si deux critères principaux et un critère secondaire du tableau 2 sont remplis. Le diagnostic différentiel doit être posé avec les véritables engelures (pernions), la sarcoïdose (lupus pernio), la thrombose et, plus rarement, la cryoglobulinémie, la macroglobulinémie ou les vascularites.

Dermatomyosite
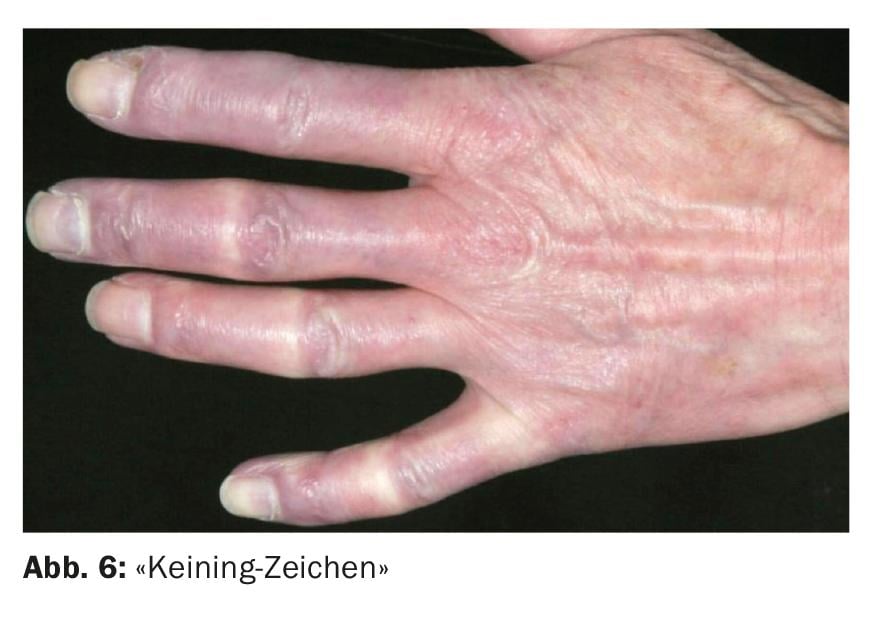
Les patients atteints de dermatomyosite peuvent développer des symptômes cutanés caractéristiques au niveau des mains et des pieds. Des papules rougeâtres, d’un blanc porcelainé, apparaissent souvent sur la face dorsale des doigts (“papules de Gottron”) (figure 5). Au niveau périunguéal, on observe souvent des érythèmes avec des télangiectasies et des hémorragies en écharde, ainsi que des hyperkératoses de l’éponychium, dont le recul est ressenti comme douloureux et est appelé “signe de Keining” (fig. 6). En outre, les ongles peuvent avoir un aspect terne et strié. La microscopie capillaire révèle principalement des ectasies capillaires et des hémorragies, sans qu’il y ait de raréfaction des vaisseaux. L’hyperémie relative et le ralentissement du flux sanguin constituent le corrélat microscopique de la coloration livide visible de la peau.

Maladie des tissus conjonctifs mixtes (syndrome de Sharp)
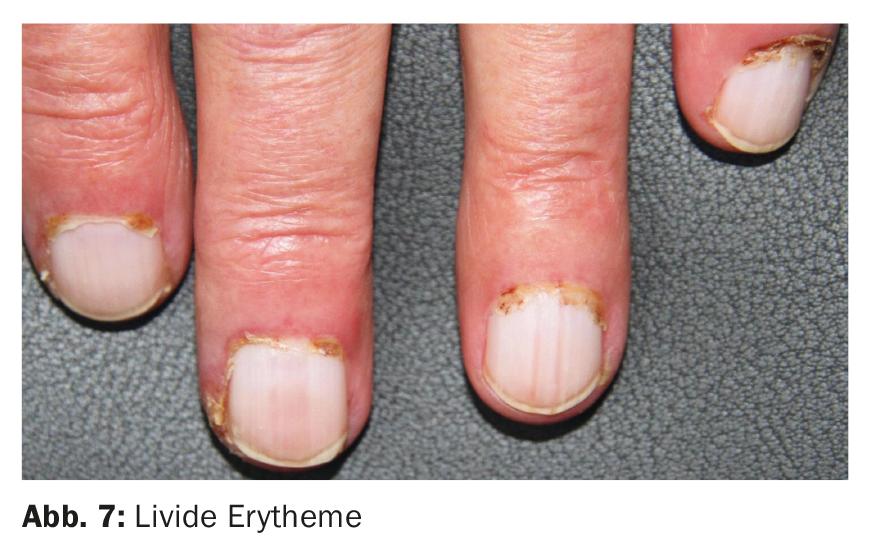
En fonction de la sévérité et des symptômes principaux, le syndrome de Sharp se caractérise par des lésions cutanées parfois spécifiques, parfois non spécifiques, au niveau des mains et des pieds. La sclérodactylie est principalement observée chez les patients plus enclins à la sclérodermie systémique. Les érythèmes périunguéaux avec télangiectasies ainsi que les érythèmes livide (figure 7) sont plutôt caractéristiques d’un lupus ou d’une dermatomyosite overlap.
Littérature complémentaire :
- Krieg T : Sclérodermie. Dermatologie et vénérologie, 5e édition, 676-689. Springer Verlag 2005.
- Meurer M : Lupus érythémateux. Dermatologie et vénérologie, 5e édition, 690-705. Springer Verlag 2005.
- Messer G : Dermatomyosite et autres maladies auto-immunes. Dermatologie et vénéréologie, 5e édition, 706-715. Springer Verlag 2005.
- Hunzelmann N, Krieg T : Sclérodermie : Dermatologie, vénérologie et allergologie, 6e édition, 849-865. Springer Verlag 2012.
- Sticherling M, Kuhn A : Lupus érythémateux Dermatologie, vénérologie et allergologie, 6e édition, 866-882. Springer Verlag 2012.
- Eming R : Dermatomyosite et autres maladies auto-immunes. Dermatologie, vénérologie et allergologie, 6e édition, 883-896. Springer Verlag 2012.
- Hunzelmann N : Sclérodermie systémique. Le dermatologue 2013 ; 64 : 299-312.
- Persa OD, et al : Sclérodermie systémique. Der Hautarzt 2015 ; 66 : 599-603.
- Volc-Platzer B : Dermatomyosite – mise à jour. Le dermatologue 2015 ; 66 : 604-610.
- Günther C, Beisserter S : Lupus érythémateux. Le dermatologue 2015 ; 66 : 611-616.
- L’encyclopédie en ligne d’Altmeyer. (www.enzyklopaedie-dermatologie.de)
- Sander S, et al. : Atlas de poche de la microscopie capillaire, 3e édition, 2008.
DERMATOLOGIE PRATIQUE 2022 ; 32(4) : 10-13