Ce syndrome très rare est dû à des mutations du gène LRP2codant pour la mégaline et se transmet sur le mode autosomique récessif. Il se caractérise par des signes craniofaciaux avec des traits faciaux caractéristiques. Outre les complications oculaires et la surdité neurosensorielle, d’autres anomalies cliniques sont décrites. Le diagnostic de suspicion peut être confirmé par des tests génétiques. Selon l’évolution individuelle, différentes mesures thérapeutiques sont nécessaires.
Le syndrome de Donnai-Barrow (DBS) est associé à de multiples malformations congénitales [1]. Les personnes atteintes se caractérisent par une dysmorphie faciale typique, une myopie et d’autres anomalies oculaires, ainsi qu’une surdité, une agénésie du corps calleux, une protéinurie de faible poids moléculaire et différents troubles du développement intellectuel. Une hernie diaphragmatique congénitale et/ou une omphalocèle sont fréquentes. Il existe peu de données sur la prévalence et l’incidence du DBS. Jusqu’à présent, moins de 50 personnes atteintes ont été décrites dans une vingtaine de familles. Le DBS est présent dans toutes les ethnies et il ne semble pas y avoir de gradient de sexe. Des mutations dans le gène LRP2 (low-density lipoprotein receptor-related protein 2 ; 2q31.1) sont à l’origine du DBS. Ce gène code pour la mégaline, qui est exprimée dans plusieurs épithéliums de résorption, notamment dans le cerveau, les reins et les yeux. La mégaline joue un rôle important dans différentes chaînes de signalisation et dans l’endocytose de nombreux ligands.
Caractéristiques cliniques fréquentes
Presque tous les patients présentent les symptômes suivants [1] :
- Agénésie/hypogénésie du corps calleux
- fontanelle antérieure hypertrophiée
- surdité neurosensorielle sévère (c’est-à-dire que le son atteint l’oreille interne, mais ne peut pas être converti en impulsions nerveuses ou n’est pas transmis au cerveau)
- Hypertélorisme.
Les traits caractéristiques du visage sont [1] :
- fentes palpébrales obliques vers le bas,
- nez court avec une arête nasale plate,
- front large et haut
- “widow’s peak” dans la ligne capillaire antérieure et parfois proptosis
Environ 40% des patients présentent une hernie diaphragmatique congénitale et/ou une omphalocèle. Souvent, le développement est retardé et l’intelligence est réduite à des degrés divers. Une myopie sévère (>6 dptr) peut entraîner un décollement/une dystrophie de la rétine et une baisse progressive de l’acuité visuelle. Des cas de colobome de l’iris, de glomérulosclérose segmentaire focale et de dysfonctionnement tubulaire proximal (rarement jusqu’à l’insuffisance rénale) ont été occasionnellement rapportés.
| Diagnostic et DD Le diagnostic du syndrome de Donnai-Barrow (DBS) repose sur une combinaison de signes cliniques et d’imagerie, ainsi que sur un schéma typique de protéinurie de faible poids moléculaire, de taux urinaires élevés de protéine de liaison au rétinol (RBP) et de rapport RBP/créatinine. Le diagnostic est confirmé par l’analyse de l’ADN. En raison de la constellation caractéristique de symptômes, le nombre de diagnostics différentiels du DBS est limité. Certains symptômes communs incluent la tétrasomie 12p, ainsi que les syndromes de Fryns, de Chudley-McCullough, acrocallosal et cranio-fronto-nasal. Le phénotype rénal ressemble en partie à la maladie de Dent et au syndrome de Lowe. Le phénotype oculaire peut évoquer le syndrome de Stickler. La mise en évidence d’un hypertélorisme et d’une hernie diaphragmatique congénitale ou d’une omphalocèle à l’imagerie prénatale doit faire penser au DBS. Le diagnostic prénatal dans les grossesses à risque nécessite l’identification préalable des mutations à l’origine de la maladie dans la famille. |
| d’après [1] |
Gestion thérapeutique et pronostic
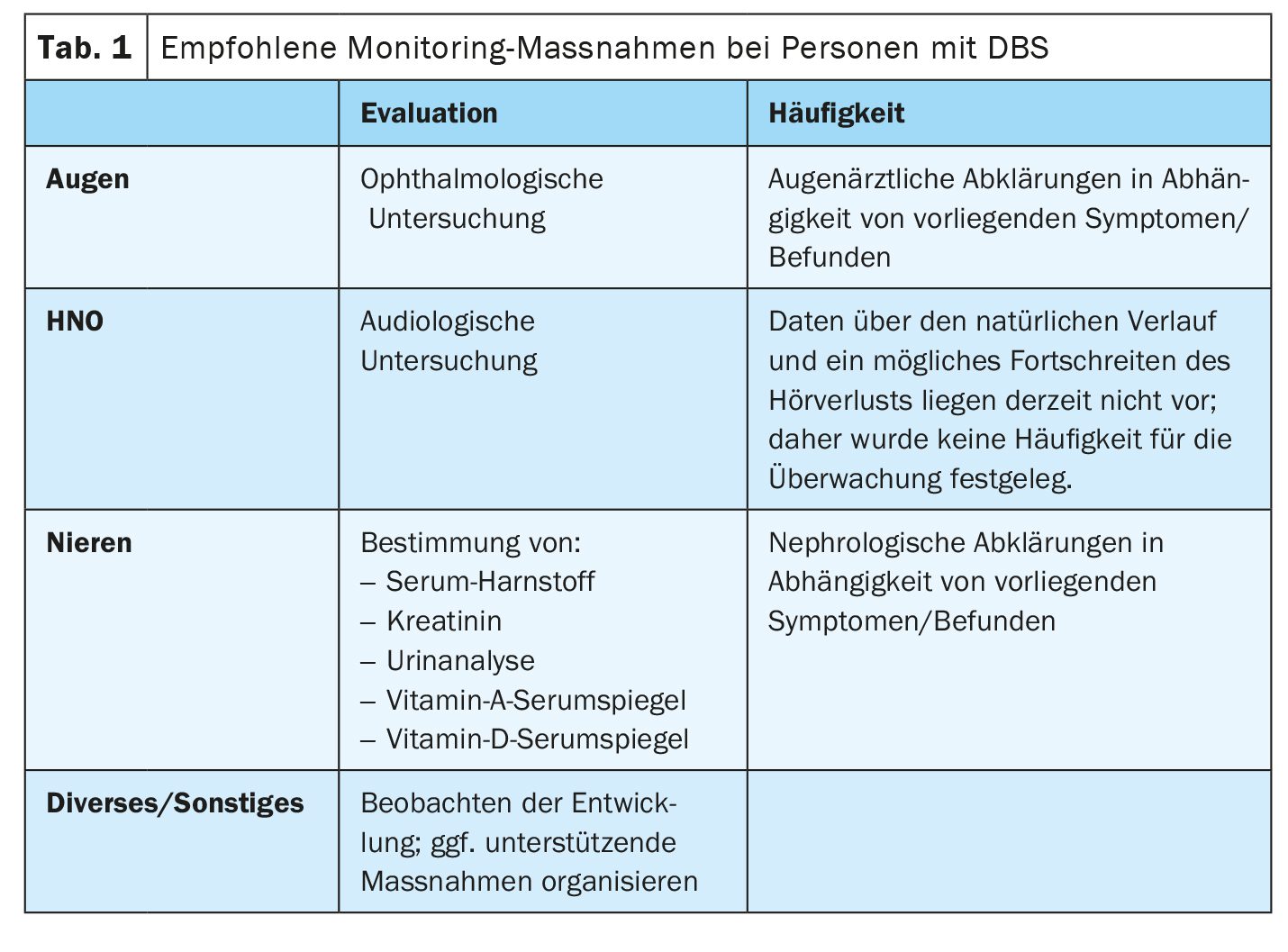
Le SCD se transmet selon un mode autosomique récessif. Un conseil génétique doit être proposé aux parents des enfants atteints et à leurs proches [1]. A l’exception d’un seul cas de disomie uniparentale publié, les parents des patients dont les cas ont été documentés se sont révélés être obligatoirement hétérozygotes. Des contrôles réguliers de l’acuité visuelle, de l’audition et de la fonction rénale sont nécessaires. La correction des lunettes, le traitement pour prévenir le décollement de la rétine, les aides auditives et/ou les implants cochléaires font partie du plan de traitement. La hernie diaphragmatique congénitale et/ou l’omphalocèle peuvent nécessiter une intervention chirurgicale. Des mesures de soutien spécifiques doivent être proposées aux enfants concernés. Les personnes concernées peuvent obtenir une vision et une audition utilisables avec une correction. L’état de santé général des patients est généralement bon pendant l’enfance et l’adolescence. L’insuffisance rénale au stade terminal est une complication rare qui peut mettre la vie en danger. La présentation pré- ou périnatale avec des défauts du diaphragme et de la paroi abdominale nécessite une intervention chirurgicale et est associée à une augmentation de la morbidité et de la mortalité.
Étude de cas : évolution de la naissance à l’école primaire
Un garçon actuellement âgé de 9 ans est né de parents caucasiens en bonne santé, sans lien de parenté, alors âgés de 34 ans (pour la mère) et de 40 ans (pour le père) [2]. Il avait une sœur et deux demi-frères et sœurs maternels en bonne santé. Pendant la grossesse, une petite exomphalose a été détectée par échographie. Le patient a été mis au monde par un accouchement vaginal normal. Les examens cliniques et d’imagerie postnatale ont révélé un hypertélorisme prononcé, un colobome bilatéral (formation d’une fente au niveau de l’œil), l’absence de corps calleux, une malrotation intestinale, des hernies inguinales bilatérales, mais pas de hernie diaphragmatique congénitale.
Omphalocèle : La hernie ombilicale a été réduite le premier jour, ses hernies inguinales ont été réparées à l’âge de 1 an et la malrotation a été opérée définitivement à l’âge de 18 mois.
Agénésie du corps calleux : à l’âge de 4 mois, le périmètre crânien du patient était de 44,5 cm (95e percentile), sa taille de 60,6 cm (90e percentile) et son poids de 5,78 kg (90e percentile). L’IRM du cerveau a confirmé l’agénésie du corps calleux et a également révélé une encéphalocèle frontale avec une fosse antérieure élargie et une malformation de Chiari 1 avec des amygdales cérébelleuses s’étendant jusqu’à C1.
Manifestations oculaires : En plus des colobomes iriens et choriorétiniens bilatéraux, il y avait une myopie élevée, une cataracte inférieure droite et un lenticône postérieur gauche, diagnostiqués à l’âge de 3 mois. La myopie était associée à des globes oculaires hypertrophiés (longueur axiale de 30 mm à l’âge de 7 ans) et à des staphylomes postérieurs bilatéraux, et il a subi une rétinopexie prophylactique au laser à 360 degrés pour éviter un décollement de la rétine. Sa prescription de lunettes était OD (Oculus Dexter) -15,00 D, OS (Oculus Sinister) -19,25/-2,00 axe 92°, bien qu’il portât normalement des lentilles de contact et atteignît une acuité visuelle corrigée de OD 20/200, OS 20/100. A l’âge de 7 ans, le patient a présenté des résultats normaux à l’électrodiagnostic, mais a signalé une baisse de la vision, en particulier la nuit, au cours des deux dernières années. Une nouvelle électrorétinographie réalisée à l’âge de 9 ans a révélé un dysfonctionnement rétinien généralisé affectant à la fois le système des bâtonnets et celui des cônes, principalement à l’interface entre le photorécepteur et l’épithélium pigmentaire rétinien. Les mesures oculaires à l’âge de 6 ans étaient respectivement de 45, 70 et 110 mm pour la distance canthal interne, la distance pupillaire et la distance canthal externe.
Résultats audiologiques : en raison d’une surdité bilatérale sévère, le patient a reçu des implants cochléaires à l’âge de 4 ans, qui ont été révisés à l’âge de 6 ans et à l’âge de 8 ans.
Développement scolaire : le patient fréquente une école ordinaire et reçoit un soutien spécial pour ses déficits visuels et auditifs. Il présente un certain retard de développement et fréquente une classe dans laquelle il a deux ans de retard sur ses camarades. Cependant, il progresse bien dans cette classe et on pense qu’une grande partie de son retard de développement est due à son déficit bisensoriel et aux lacunes dans sa scolarité dues à ses fréquents séjours à l’hôpital.
Confirmation du diagnostic : le diagnostic de suspicion de DBS a été confirmé par un test génétique qui a révélé une délétion homozygote de 4 pb (c.11469_11472delTTTG) dans l’exon 60 du gène LRP2 par séquençage direct.
Littérature :
- “Syndrome de Donnai-Barrow”, www.orpha.net,(dernière consultation 27.09.2024).
- Kantarci S, et al : Syndrome de Donnai-Barrow (DBS/FOAR) chez un enfant avec une mutation homozygote LRP2 due à une isodisomie paternelle complète du chromosome 2. Am J Med Genet A 2008 ; 146A(14) : 1842-1847.
- Longoni M, et al. : Syndrome de Donnai-Barrow. 2008 Aug 28 [Updated 2018 Nov 21]. In : Adam MP, et al. (Eds). GeneReviews® [Internet]. Seattle (WA) : Université de Washington, Seattle ; 1993-2024.
PRATIQUE DU MÉDECIN DE FAMILLE 2024 ; 19(10) : 24-25