
Le syndrome de Fanconi-Bickel (FBS) se produit en raison de variantes du gène SLC2A2. Le diagnostic d’une maladie génétique rare peut prendre jusqu’à 5-6 ans, voire plus dans les pays à revenu faible ou intermédiaire où les ressources technologiques sont limitées. Des médecins péruviens ont présenté le cas d’un enfant de deux ans et demi présentant des troubles de la croissance, une hépatomégalie, une acidose métabolique, une hypophosphatémie, une hypokaliémie et une hyperlactatémie.
Le syndrome de Fanconi-Bickel (OMIM #227810), une maladie héréditaire autosomique récessive (AR), est caractérisé par une combinaison de maladies hépatiques et rénales causées par un défaut du transporteur de glucose GLUT2 (gène SLC2A2). Cela entraîne une accumulation de glycogène, un dysfonctionnement tubulaire rénal proximal et une altération de l’utilisation du glucose et du galactose.
Le phénotype comprend un manque de prise de poids, un ventre gonflé, une hépatomégalie, une hypoglycémie à jeun, une hyperglycémie postprandiale, une glucosurie, une phosphaturie, une aminoacidurie, une polyurie, une acidose métabolique, une ostéoporose, une hypophosphatémie, un rachitisme et la présence de glycogène dans la biopsie hépatique ou rénale. Dans de rares cas, un carcinome hépatocellulaire a été observé en raison de l’activation de la voie de signalisation Wnt. Cependant, des cas de patients présentant des symptômes cliniques légers ont été signalés, y compris des cas de glucosurie pure. Le gène SLC2A2 (OMIM *138160) contient 11 exons et sa protéine GLUT2 est composée de 524 acides aminés et se trouve dans la membrane cellulaire, exprimée dans les hépatocytes, les entérocytes, les tubules rénaux proximaux, les cellules bêta du pancréas, les neurones et les astrocytes. Les variantes pathogènes de SLC2A2 modifient l’entrée et la sortie du glucose dans les hépatocytes et réduisent la sécrétion d’insuline en raison d’une sensibilité accrue des cellules bêta en phase postprandiale.
Rapport de cas d’un garçon de 2½ ans
Un garçon âgé de 2 ans et 7 mois, né et élevé au Pérou de la cinquième grossesse de parents apparentés, a été présenté à l’équipe du Dr Hernán Abarca-Barriga de l’Institut national de la santé (INS) du Pérou pour vomissements, diarrhée, acidose métabolique, hypokaliémie et hyperlactatémie, hypoactivité et fièvre. Il avait déjà été hospitalisé pour ces symptômes à l’âge de 1 an et 10 mois et de 2 ans et 2 mois [1].
L’enfant est né avec un poids de naissance de 3620 g, une taille de 49 cm et un périmètre crânien de 34 cm (percentile normal), et un score d’Apgar de 8-9. En ce qui concerne le développement psychomoteur, il a atteint le contrôle de la tête à un mois, s’est assis sans assistance à sept mois, a marché avec assistance à un an et six mois. Il a prononcé ses premiers mots à un an et cinq mois, a dit deux mots à deux ans et neuf mois et a montré un sourire social à un an. Il a été examiné à l’âge d’un an et huit mois en raison d’un poids insuffisant et d’une croissance réduite, d’une diarrhée chronique, d’une fièvre et d’une augmentation du volume abdominal (Figure 1).
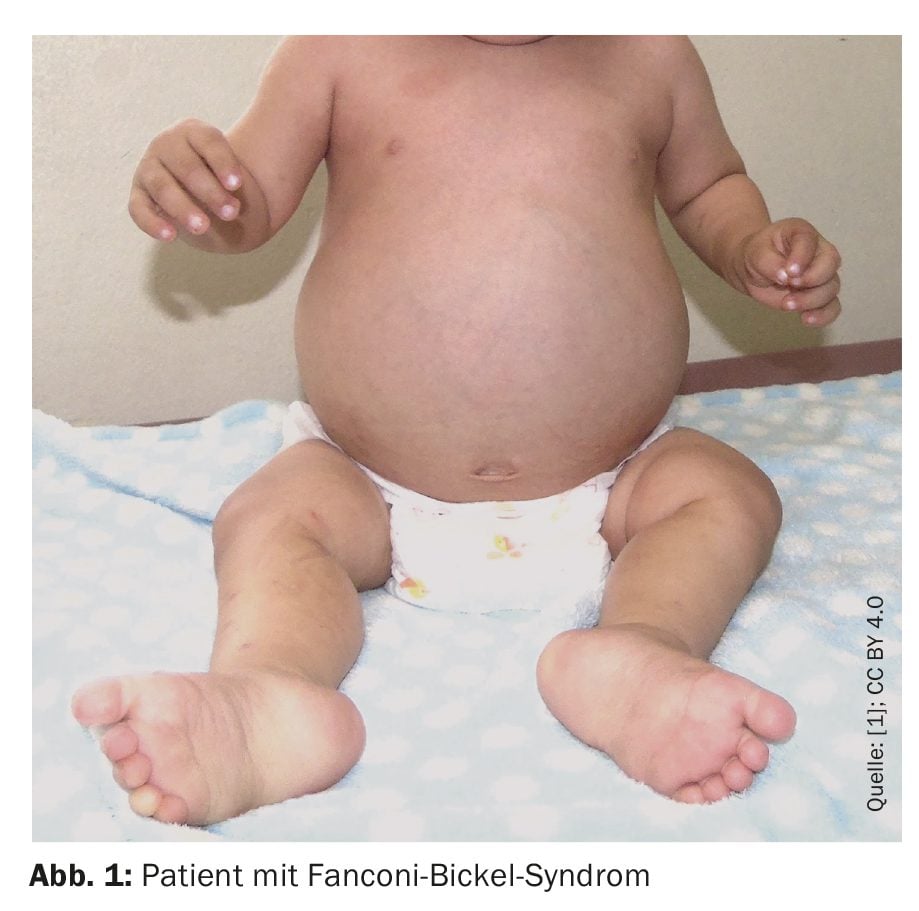
Lors de l’admission, les médecins ont constaté une protubérance du front, une hépatomégalie, une hypotonie et une déformation pseudo-Madelung. Le poids et la taille de l’enfant étaient inférieurs au premier centile depuis l’âge de six mois, tandis que son périmètre crânien était dans la norme. Les radiographies ont montré un effilochage et un élargissement des métaphyses du fémur (distal) et du tibia (proximal), ce qui est compatible avec le rachitisme.
Les résultats cliniques et de laboratoire ont permis de conclure à un SFC
Le petit patient présentait une hypoglycémie, une hypocalcémie et une hypercalcémie, une hypophosphatémie, une hypercholestérolémie, une hypertriglycéridémie, une hyperphosphatasémie, une hypokaliémie en cas de diarrhée et de vomissements, une hyperlactatémie et une hypouricémie. L’analyse d’urine a montré des valeurs normales de pH et de HCO3, mais une hyperprotéinurie, une hypocréatinurie, une microalbuminurie, une hyperglycosurie et une hypercalciurie. En outre, le garçon présentait une thrombocytose. Le gaz veineux présentait un pH de 7,261-7,5 mmHg, un HCO3 de 7,5-28,8 mmHg et un excès de base de -15,8 à +5,3. Ces analyses ont permis de diagnostiquer une acidose tubulaire rénale. L’échographie de l’abdomen a montré une hépatomégalie, il n’y avait aucun signe de fibrose ou de néphromégalie.
Une biopsie du foie a révélé une architecture hépatique partiellement déformée en raison de la présence de quelques hypertrophies fibreuses de l’espace porte, d’une infiltration inflammatoire de lymphocytes, d’hépatocytes de grande taille et ballonnés avec un motif en mosaïque et une légère fibrose péricellulaire. La coloration de Schiff à l’acide périodique avec diastase met en évidence les dépôts d’éosinophiles dans les hépatocytes, qui sont corrélés aux dépôts de glycogène. Sur la base de ces résultats cliniques et de laboratoire, les médecins ont suspecté un syndrome de Fanconi-Bickel (FBS).
Séquençage de l’exome identifiant le variant pathogène homozygote
Des résultats de tests génétiques ont été obtenus à l’âge de 1 an et 8 mois en utilisant l’ADN génomique. Au total, 131 477 variants annotés ont été identifiés dans 18 179 gènes, en excluant les variants qui étaient probablement bénins ou légers. Pour identifier les variants associés au phénotype du patient, les termes “trouble de la croissance” (HPO : 0001508) et “hépatomégalie” (HPO : 0002240) ont été utilisés, les auteurs précisant qu’un seuil de fréquence allélique de population de 1% a été pris en compte. En raison de l’hypophosphatémie, du rachitisme et du stockage de glycogène dans le foie, les chercheurs ont également recherché manuellement des variants dans SLC2A2. En raison de la consanguinité parentale, la priorité a été donnée à l’analyse des homozygotes. Une fréquence allélique des variants (VAF) supérieure à 0,9 a été utilisée pour sélectionner les gènes candidats potentiels. Le séquençage de l’exome a identifié un variant homozygote non sens dans le gène SLC2A2, qui a été décrit comme pathogène.
Le diagnostic clinique du patient était basé sur la présence d’une hépatomégalie, d’une hypoglycémie, d’une glucosurie, d’une hypophosphatémie, d’une hyperphosphatasie, d’une hypouricémie, d’un rachitisme, d’une pseudo-déformation de Madelung et d’une acidose tubulaire rénale. La présence d’une aminoacidurie n’a toutefois pas pu être établie, car le test n’était pas disponible sur place. Cette excrétion insuffisante d’acides aminés aurait facilité le diagnostic clinico-biochimique, expliquent les auteurs. La confirmation moléculaire en utilisant le séquençage Sanger du gène SLCA2 a été un autre obstacle, car ce test n’est pas disponible au Pérou. Compte tenu de l’accès limité aux tests génétiques dans le pays, il n’a pas été possible de confirmer la présence de la variante chez les parents au premier degré du patient, soulignent les auteurs. Dans ce cas, le séquençage de l’exome a permis aux chercheurs d’identifier précisément le variant homozygote.
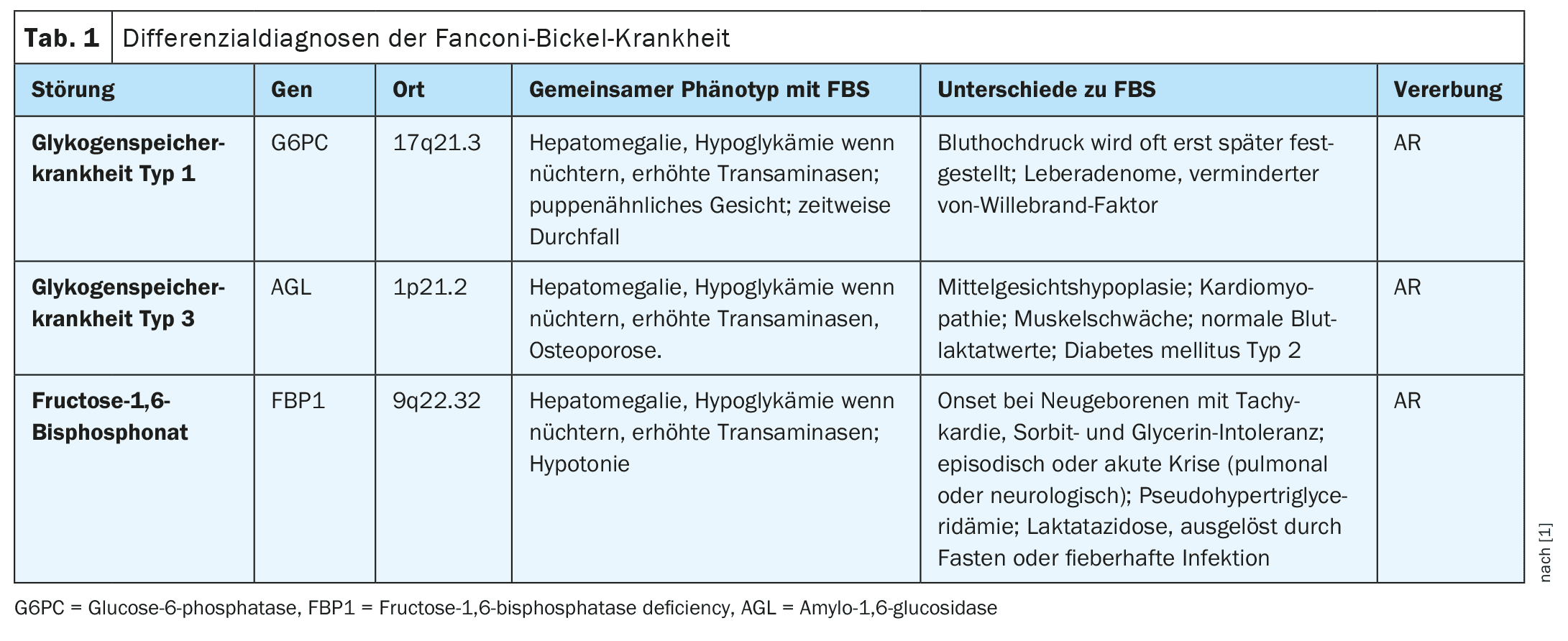
Selon le Dr Abarca-Barriga et ses collègues, l’augmentation du lactate sanguin observée pourrait être due à une charge accrue en glucose due au métabolisme anaérobie. En outre, un faible taux d’acide urique dû à un dysfonctionnement du tubule proximal fait partie du phénotype des patients atteints de SFC, ce qui entraîne une hyperuricosurie. Certaines caractéristiques cliniques, telles qu’un retard de développement du langage, sont également probablement associées à la présence d’une hypoglycémie chronique.
Il n’existe à ce jour aucun traitement causal pour le syndrome de Fanconi-Bickel. Le jeune patient a été traité avec du bicarbonate, de l’amlodipine, du citrate de sodium et une solution d’acide citrique, de l’énalapril, de l’alendronate et du zolendronate, et a reçu un traitement nutritionnel à base d’amidon de maïs non cuit, ce qui a entraîné une amélioration du poids et de la taille. En outre, les auteurs soulignent que depuis le début du traitement diététique à l’amidon de maïs non cuit, le patient a montré une amélioration d’un écart-type (ET) en termes de poids et de taille. L’utilisation de l’amidon de maïs est donc essentielle non seulement pour prévenir l’hypoglycémie nocturne, mais aussi pour améliorer la taille et le poids des patients.
Littérature :
- Abarca-Barriga HH, et al.: Importance about use of high-throughput sequencing in pediatric: case report of a patient with Fanconi-Bickel syndrome. BMC Pediatr 2024; 24: 161; doi: 10.1186/s12887-024-04641-1.
HAUSARZT PRAXIS 2024; 19(11): 48–49









