En cas de suspicion de tumeur sécrétant des catécholamines, le dosage des métanéphrines dans le plasma libre est une méthode de diagnostic sensible. L’hypertension paroxystique est un symptôme fréquent. En fonction des résultats de laboratoire, un examen d’imagerie doit être effectué afin de localiser la tumeur. Pour les tumeurs non résécables ou métastatiques, il existe de nouvelles thérapies expérimentales basées sur la signature génétique et moléculaire individuelle, dans lesquelles on place beaucoup d’espoir. Dans ce contexte, les analyses génétiques gagnent en importance.
Les phéochromocytomes (PCC) sont des tumeurs productrices de catécholamines qui se développent à partir des cellules chromaffines de la médullosurrénale. Les paragangliomes (PGL) sont des tumeurs produisant de la noradrénaline et de la dopamine dans les paraganglions sympathiques et parasympathiques [1]. Le tableau clinique et la procédure diagnostique initiale sont très similaires pour les deux types de tumeurs sécrétant des catécholamines [2]. Un symptôme typique du phéochromocytome est l’hypertension, qui survient de manière paroxystique chez environ la moitié des patients atteints. Les autres signes de la maladie sont par exemple la transpiration, les palpitations, les maux de tête, les tremblements ou la nausée. Dans la plupart des cas, les PGL sont facilement guérissables par résection complète, mais il existe des différences considérables dans le risque de malignité en fonction du génotype et du locus tumoral. Les tumeurs considérées comme malignes sont celles qui métastasent, soit environ 10 à 15% de tous les CPC et 35 à 40% de tous les PGL [1]. Svenja Nölting, médecin-chef à la Clinique d’endocrinologie, de diabétologie et de nutrition clinique de l’Hôpital universitaire de Zurich, a souligné que tous les patients atteints de phéochromocytomes/paragangliomes (PPGL) devraient faire l’objet d’une analyse génétique lors de la réunion annuelle de la Société suisse d’endocrinologie et de diabétologie (SSED) en novembre dernier [1].

Typage des clusters : signature génétique et moléculaire
“Environ 70% de tous les PPGL peuvent être attribués à l’un des trois clusters moléculaires en fonction de leur mutation sous-jacente”, a expliqué la conférencière [1,3]. La plupart des tumeurs appartiennent aux clusters 1 et 2, mais on sait encore peu de choses sur le cluster 3. Les tumeurs de cluster 1 sont basées sur l’activation de voies de signalisation pseudohypoxiques, les tumeurs de cluster 2 sur l’activation de voies de signalisation dépendantes de la tyrosine kinase [4]. Dans 30-35% des cas, il existe des mutations germinales à transmission autosomique dominante, 35-40% sont dues à des mutations somatiques. Les tumeurs de cluster 1 sont plus souvent localisées au niveau extra-surrénal et présentent le risque le plus élevé de métastases, tandis que les tumeurs de cluster 2 sont généralement localisées au niveau surrénal et présentent un faible risque de métastases [4].
Diagnostic : les métanéphrines dans le plasma libre sont des marqueurs pertinents
Les patients avec une mutation germinale connue, mais aussi en cas de suspicion clinique de phéochromocytome/paragangliome (PPGL) ou si un incidentalome surrénalien ou un PPGL a été rapporté dans l’histoire, doivent être dépistés [4]. Dans un premier temps, les paramètres biochimiques de laboratoire suivants doivent être recueillis [1] :
- Métanéphrines dans le plasma libre (=métabolites de l’adrénaline)
- Normétanéphrines (= métabolites de la noradrénaline)
- 3-méthoxytyramine (=métabolites de la dopamine)
Il est recommandé de le faire par spectrométrie de masse, car cela permet d’obtenir une meilleure sensibilité que les autres méthodes. Avant la prise de sang, les patients doivent notamment éviter la nicotine et le café, ainsi que la prise d’ISRS et/ou d’antidépresseurs tricycliques, de crystal meth ou de cocaïne. En plus des paramètres mentionnés, il est également utile de déterminer la chromogranine A, car c’est un bon marqueur, en particulier pour les tumeurs asymptomatiques, et dans ce cas, il faut éviter de prendre des inhibiteurs de la pompe à protons (IPP) une semaine avant l’examen. Le seul dosage de la catécholine n’est pas utile, selon l’intervenante, car cela peut conduire à un faux négatif. Si les taux de métanéphrine déterminés sont supérieurs au double de la norme, une imagerie est indiquée dans une étape ultérieure de l’évaluation [1]. Dans un premier temps, il est recommandé de réaliser une IRM/TDM de l’abdomen et de la région pelvienne. Si le résultat est négatif ou s’il existe déjà une tumeur extra-adrénale/PGL, une imagerie fonctionnelle est nécessaire. Si la taille de la tumeur surrénalienne est <5 cm, le Pr Nölting conseille d’utiliser le DOPA PET/CT. La TEP/TDM DOTA-SSA doit être réalisée en présence de tumeurs extra-surrénaliennes et d’une imagerie anatomique négative, ou si des métastases sont déjà présentes, ou si la taille de la tumeur surrénalienne est >5 cm, a expliqué l’oratrice [1].
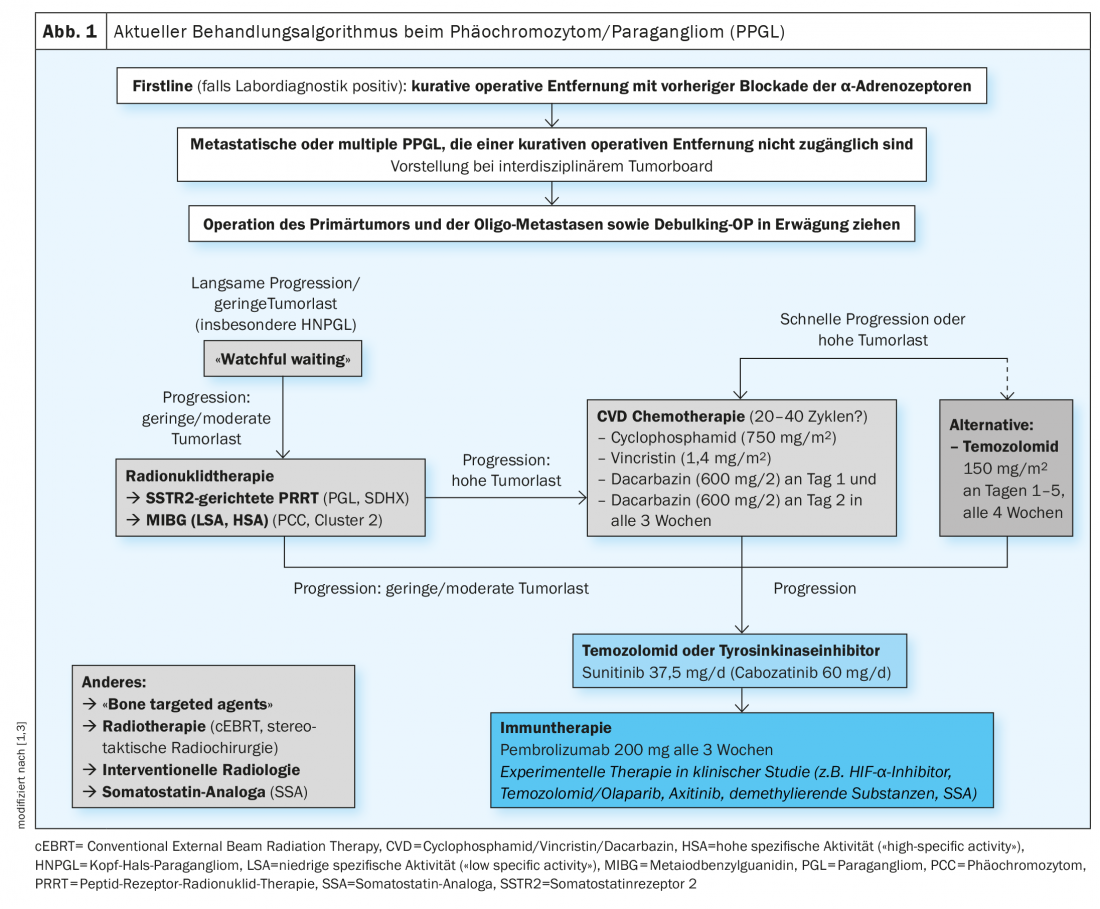
Stratégie de traitement individualisée : nouvelles thérapies expérimentales
L’algorithme de traitement actuel est présenté dans la figure 1 [1,3]. Le traitement de première ligne des PPGL localisées est l’ablation chirurgicale. Cette opération peut généralement être réalisée de manière peu invasive et doit être effectuée dans un centre expérimenté [4]. Pour éviter les complications, un blocage médicamenteux des récepteurs α-adrénergiques est effectué en préopératoire. Dans le cas des PPGL métastatiques, le traitement actuel fait appel à la radionucléothérapie, à la chimiothérapie ou aux inhibiteurs de tyrosine kinase, même si aucun traitement n’a encore été officiellement approuvé. D’autres nouvelles stratégies de traitement sont en cours d’évaluation clinique. Jusqu’à présent, le traitement spécifique aux clusters de la maladie inopérable ou métastatique n’est pas encore établi dans la pratique clinique quotidienne. Cela serait pourtant utile pour pouvoir proposer un traitement personnalisé, guidé par la génétique, selon le professeur Nölting [3]. Une publication scientifique parue dans l’Endocrine Reviews en 2021 explique en détail comment élaborer un plan de prise en charge cohérent et individualisé pour les patients atteints de phéochromocytome/paragangliome sur la base d’analyses génétiques ou d’un typage en grappes afin d’adapter au mieux les options de traitement [3].
Congrès : Société suisse d’endocrinologie et de diabétologie 11.11.2021
Littérature :
- Nölting S : “Genetic testing in patients with pheochromocytomas/paragangliomas”, Prof. Dr med. Svenja Nölting, SSED 11.11.2021
- Zulewski H, Grouzmann E : Diagnostic et traitement : “Phéochromocytome”, Swiss Med Forum 2017 ; 17(37) : 790-796, https://medicalforum.ch/de/detail/doi/smf.2017.03057
- Nölting S, et al : Gestion personnalisée du phéochromocytome et du paragangliome. Endocr Rev 2021 Jun 19:bnab019
- Phéochromocytome – maladie modèle pour la médecine personnalisée, Dtsch Med Wochenschr 2021 ; 146(23) : 1520-1526.
- Société allemande d’endocrinologie, d’hormones et de métabolisme : PROSPHEO, www.endokrinologie.net/sektion-nebenniere-steroide-hypertonie-7.php (dernière consultation 07.12.2021)
PRATIQUE DU MÉDECIN DE FAMILLE 2022 ; 17(1) : 42-43
InFo ONKOLOGIE & HÉMATOLOGIE 2022 ; 10(3) : 18-19