
Le pronostic de cette maladie autosomique récessive dépend d’un diagnostic posé dès la période néonatale et du début du traitement qui s’ensuit, notamment en ce qui concerne la survenue de complications telles que le carcinome hépatocellulaire et les troubles neurocognitifs. Le traitement pharmacologique actuel est la nitisinone, mais certaines mesures diététiques sont également recommandées.
Dans la tyrosinémie hépatorénale de type 1 (HT1), la formation de l’enzyme fumarylacétoacétase (FAH), la dernière enzyme de la dégradation de la tyrosine, est perturbée dès la naissance. Cela est dû à une variante pathogène biallélique dans le gène FAH. En raison de la carence enzymatique, les métabolites toxiques que sont l’acétoacétate de fumaryle, l’acétoacétate de maléyle et l’acétone succinique s’accumulent dans l’organisme et endommagent le foie, les reins et le système nerveux périphérique. Par la suite, en l’absence de traitement, une insuffisance hépatique survient souvent dès la première année de vie ou, si l’évolution est plus lente et chronique, une cirrhose du foie ou un carcinome hépatocellulaire (CHC) se développent [1]. La nitisinone est une substance active qui intervient dans la cascade de dégradation précoce de la tyrosine en inhibant de manière compétitive l’enzyme 4-hydroxyphénylpyruvate dioxygénase. Cela permet d’éviter la formation d’intermédiaires toxiques. Le traitement de tous les génotypes de la maladie doit être initié le plus tôt possible afin de prolonger la survie globale et de prévenir les manifestations organiques mettant en jeu le pronostic vital. En Suisse, la nitisinone (Nityr®) a été autorisée en 2022 sous forme de comprimés [2]. Le début du traitement dans les premières semaines de vie permet non seulement de prévenir le CHC dans la plupart des cas, mais aussi d’éviter les troubles hépatiques et rénaux [3–5].
Avant l’ère de la nitisinone, l’évolution naturelle de l’HT1 était généralement fatale, 90% des patients HT1 mouraient dans les deux premières années de leur vie et la seule option était la transplantation hépatique [1]. La disponibilité de la nitisinone (NTBC) a donc fondamentalement modifié l’évolution clinique et le résultat des personnes atteintes de HT1, selon les auteurs du guide S2k sur le diagnostic et le traitement de la tyrosinémie hépatorénale (tyrosinémie de type 1), mis à jour en 2022 [1].
Dépistage néonatal recommandé
Un début de traitement dès la période néonatale est décisif pour le pronostic à long terme, notamment pour la prévention d’un CHC. Comme les premiers symptômes n’apparaissent qu’à l’âge de quelques mois chez la plupart des patients, il n’est pas possible de poser un diagnostic précoce d’un point de vue purement clinique [3]. Par conséquent, en cas de suspicion clinique, la suczinylacétone (SA) doit être déterminée quantitativement à partir de sang sec, de sérum/plasma et/ou d’urine (“dépistage sélectif”). La seule détermination de la concentration de tyrosine présente une sensibilité et une spécificité insuffisantes et n’est donc pas recommandée [1]. Une mesure de SA supérieure à un cut-off défini est considérée comme un résultat de dépistage positif. Pour confirmer le diagnostic, une analyse génétique moléculaire du gène FAH peut être effectuée (encadré). Si le gène FAHest indétectable, la ligne directrice recommande d’analyser le gène GSTZ1 [1].
| Analyses de génétique moléculaire L’identification de variants pathogènes bialléliques dans le gène de la fumarylacétoacétase (FAH)confirme le diagnostic de tyrosinémie de type 1 [1]. Pour cette analyse, on peut utiliser de l’ADN provenant de cellules sanguines ou d’autres tissus corporels (frottis de la joue). Il est également possible d’analyser l’ARN à partir de cellules sanguines, d’échantillons de biopsie hépatique ou de fibroblastes en culture. L’analyse monogénique des 14 exons codants du gène FAHchez les patients présentant un tableau clinique/biochimique clair est une méthode connue de longue date qui permet de détecter des variantes pathogènes avec une sensibilité estimée à >95% [1]. Cependant, de nos jours, les techniques NGS(Next Generation Sequencing) sont généralement utilisées dans le cadre d’un diagnostic de panel ou dans le cadre d’un WES (Whole Exome Sequencing) pour l’analyse des gènes FAH– à la fois pour un diagnostic plus rapide et plus rentable – [16]. |
Commencer le traitement par nitisinone dans les premières semaines de vie
Les effets de la nitisinone reposent sur l’inhibition de l’enzyme 4-hydroxyphénylpyruvate dioxygénase, qui est impliquée dans la dégradation normale de la tyrosine (figure 1). Cela permet d’éviter la formation de métabolites toxiques. Les effets secondaires sont rares, les plus fréquemment rapportés étant des troubles de la formule sanguine, des troubles oculaires et une augmentation de la concentration de tyrosine. Une interruption ou un arrêt brutal du traitement par nitisinone peut entraîner des “crises de porphyrie” et doit être évité [1, 6-8]. La valeur thérapeutique cible pour la nitisinone n’est pas bien définie à ce jour, la littérature spécialisée indique 20-60 μM comme valeurs thérapeutiques cibles pour les concentrations de nitisinone dans le plasma [3,9–11]. Selon les auteurs des lignes directrices, des concentrations beaucoup plus faibles sont possibles sans compromettre le contrôle métabolique, mesuré par la suppression de la production de SA [1]. L’intervalle thérapeutique cible pour la concentration de tyrosine dans le sang est estimé à 200-800 μM, sans qu’il existe d’études comparatives contrôlées et randomisées [3,9–12].
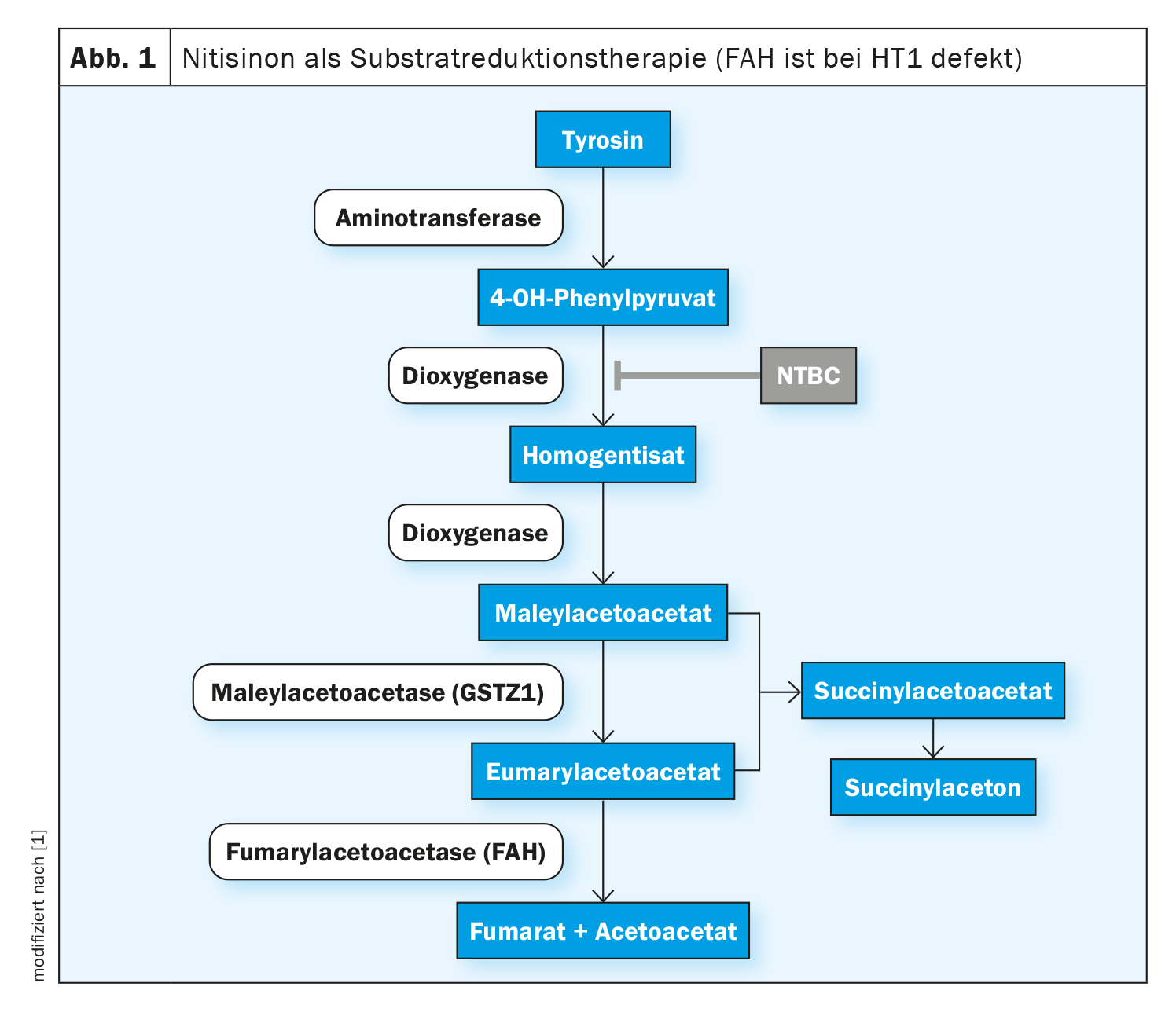
Dans la mesure du possible, le traitement par nitisinone doit être associé à un régime alimentaire réduit en protéines, supplémenté avec un mélange d’acides aminés sans tyrosine ni phénylalanine [13–15].
Les pièges du diagnostic – les analyses génétiques aident à avancer
Le principal diagnostic différentiel d’une concentration élevée de SA est le déficit en maléylacétoacétate isomérase, une anomalie métabolique probablement bénigne qui n’est pas associée à des troubles de la fonction hépatique [1]. La ligne directrice indique également que des concentrations légèrement élevées de SA – selon la valeur de cut-off utilisée – peuvent également être temporaires ou liées à des formes d’évolution bénignes ne nécessitant pas de traitement [1]. Un résultat de dépistage positif doit être confirmé par une ou plusieurs méthodes d’analyse alternatives, en plus du contrôle dans le même échantillon (sang sec). Il s’agit notamment de l’analyse quantitative par chromatographie en phase gazeuse/spectrométrie de masse (GC/MS) des acides organiques dans l’urine et de la détermination de la SA dans le sang. En cas de forte suspicion, il est également utile de vérifier les paramètres de la fonction hépatique. Les résultats anormaux du diagnostic de confirmation peuvent être clarifiés par une analyse génétique moléculaire du gène FAH. Si celle-ci est normale, une analyse du gène GSTZ1 (glutathion S-transférase Zeta 1-1), dont le déficit est à l’origine du déficit en maléylacétoacétate-isomérase, peut être effectuée [1].
Littérature :
- “S2k-Leitlinie : Diagnostik und Therapie der hepatorenalen Tyrosinämie (Tyrosinämie Typ 1)”, AWMF- numéro de registre : 027-003, mise à jour 09.06.2022.
- Swissmedic : Information sur les médicaments, www.swissmedicinfo.ch,(dernière consultation 06.02.2024)
- Mayorandan S, et al : Cross-sectional study of 168 patients with hepatorenal tyrosinaemia and implications for clinical practice. Orphanet J Rare Dis 2014 ; 9 : 107.
- McKiernan PJ, Preece MA, Chakrapani A : Outcome of children with hereditary tyrosinaemia following newborn screening. Arch Dis Child 2015 ; 100(8) : 738-741.
- Bartlett DC, et al : Le traitement précoce par nitisinone réduit le besoin de transplantation hépatique chez les enfants atteints de tyrosinémie de type 1 et améliore la fonction rénale post-transplantation. J Inherit Metab Dis 2014 ; 37(5) : 745-752.
- Önenli Mungan N, et al : Tyrosinemia type 1 and irreversible neurologic crisis after one month discontinuation of nitisone. Metab Brain Dis 2016 ; 31(5) : 1181-1183.
- Schlump JU, et al : Severe neurological crisis in a patient with hereditary tyrosinaemia type I after interruption of NTBC treatment. J Inherit Metab Dis 2008 ; 31 Suppl 2 : S223-S225.
- Uçar HK, et al : A Case Report of a Very Rare Association of Tyrosinemia type I and Pancreatitis Mimicking Neurologic Crisis of Tyrosinemia Type I. Balkan Med J 2016 ; 33(3) : 370-372.
- Chinsky JM, et al : Diagnostic et traitement de la tyrosinémie de type I : une revue du groupe de consensus américain et canadien et des recommandations. Genet Med 2017 ; 19(12) : doi:10.1038/gim.2017.101
- de Laet C, et al. : Recommandations pour la prise en charge de la tyrosinémie de type 1. Orphanet J Rare Dis 2013 ; 8 : 8. Publié en 2013 Jan 11. doi:10.1186/1750-1172-8-8
- Alvarez F, Mitchell GA : Tyrosinémie et transplantation hépatique : expérience au CHU Sainte-Justine. Adv Exp Med Biol 2017 ; 959 : 67-73.
- De Laet C, et al : Neuropsychological outcome of NTBC-treated patients with tyrosinaemia type 1. Dev Med Child Neurol 2011 ; 53(10) : 962-964.
- Morrow G, Angileri F, Tanguay RM : Aspects moléculaires des mutations FAH impliquées dans la maladie HT1. Adv Exp Med Biol 2017 ; 959 : 25-48.
- Morrow G, Tanguay RM : Aspects biochimiques et cliniques de la tyrosinémie héréditaire de type 1. Adv Exp Med Biol 2017 ; 959 : 9-21.
- van Spronsen FJ, et al. : Considérations diététiques dans la tyrosinémie de type I. Adv Exp Med Biol 2017 ; 959 : 197-204.
- Blackburn PR, et al : Silent Tyrosinemia Type I Without Elevated Tyrosine or Succinylacetone Associated with Liver Cirrhosis and Hepatocellular Carcinoma. Hum Mutat 2016 ; 37(10) : 1097-1105.
PRATIQUE DU MÉDECIN DE FAMILLE 2024 ; 19(2) : 36-37









