Les immunodéficiences primaires (IDP) se manifestent souvent dès l’enfance et peuvent entraîner des troubles majeurs jusqu’à l’âge adulte lorsqu’elles sont diagnostiquées tardivement [1]. Il existe pourtant des traitements efficaces, qui peuvent réduire la morbidité et permettre aux patients de mener une vie quasiment normale [2].
Les immunodéficiences primaires (IDP) englobent un groupe hétérogène de plus de 400 défauts génétiques, qui provoquent un dysfonctionnement du système immunitaire et se manifestent entre autres par une sensibilité pathologique aux infections ainsi que par des maladies auto-immunes [2]. En Suisse, leur prévalence est estimée entre 1:10 000 et 1:500 000, en fonction de la maladie et de la région [3]. Le nombre de cas d’IDP a toutefois fortement augmenté au cours des dernières années du fait des progrès diagnostiques, raison pour laquelle il est aujourd’hui estimé que la prévalence est nettement plus élevée [2]. Les maladies liées à un déficit en anticorps, qui résultent d’une production réduite d’anticorps et touchent environ la moitié des patients atteints d’IDP, constituent de loin le plus grand groupe d’IDP [2, 4].
Un diagnostic précoce peut réduire la mortalité
La sensibilité pathologique aux infections est un symptôme majeur de l’IDP [2]. Les manifestations cliniques sont toutefois très hétérogènes en raison de la multitude de défauts génétiques impliqués et incluent également des maladies auto-immunes et auto-inflammatoires. Cette hétérogénéité ainsi que la distinction souvent difficile avec d’autres maladies sont responsables d’une latence diagnostique considérable, avec des conséquences désastreuses: selon une étude portant sur 2 212 patients du registre de la Société européenne des immunodéficiences (ESID), le risque de décès augmente de 1,7% par année de retard diagnostique [5]. Le dépistage précoce joue donc un rôle central dans la prévention des dommages à long terme [2]. En présence d’au minimum deux des dix principaux signes d’alerte, il est judicieux de rechercher une IDP. En particulier la maladie liée à un déficit en anticorps, qui est l’IDP la plus fréquente, peut être facilement diagnostiquée au cabinet médical par la quantification des immunoglobulines IgM, IgG, IgA et IgE.
10 signes d’alerte de l’immunodéficience primaire chez les enfants
cf. Jeffrey Model Foundation: http://downloads.info4pi.org/pdfs/10-Warning-Signs—Generic-Text–2-.pdf |
Options thérapeutiques en cas d’IDP
Le traitement de l’IDP est fonction de la pathogenèse sous-jacente (aperçu dans le Tableau 1). Les maladies liées à un déficit en anticorps, qui s’accompagnent souvent d’une sensibilité pathologique aux infections, peuvent être bien traitées par un traitement de substitution des immunoglobulines (Ig): ce dernier compense le déficit en anticorps et réduit ainsi la fréquence et la sévérité des infections. Les préparations d’Ig sont purifiées à partir de plasma provenant de 10 000 à 60 000 donneurs sains et elles sont injectées aux patients soit par voie intraveineuse, soit par voie sous-cutanée [3].
Tableau 1: Aperçu des stratégies thérapeutiques en cas d’IDP. Adapté d’après [6]
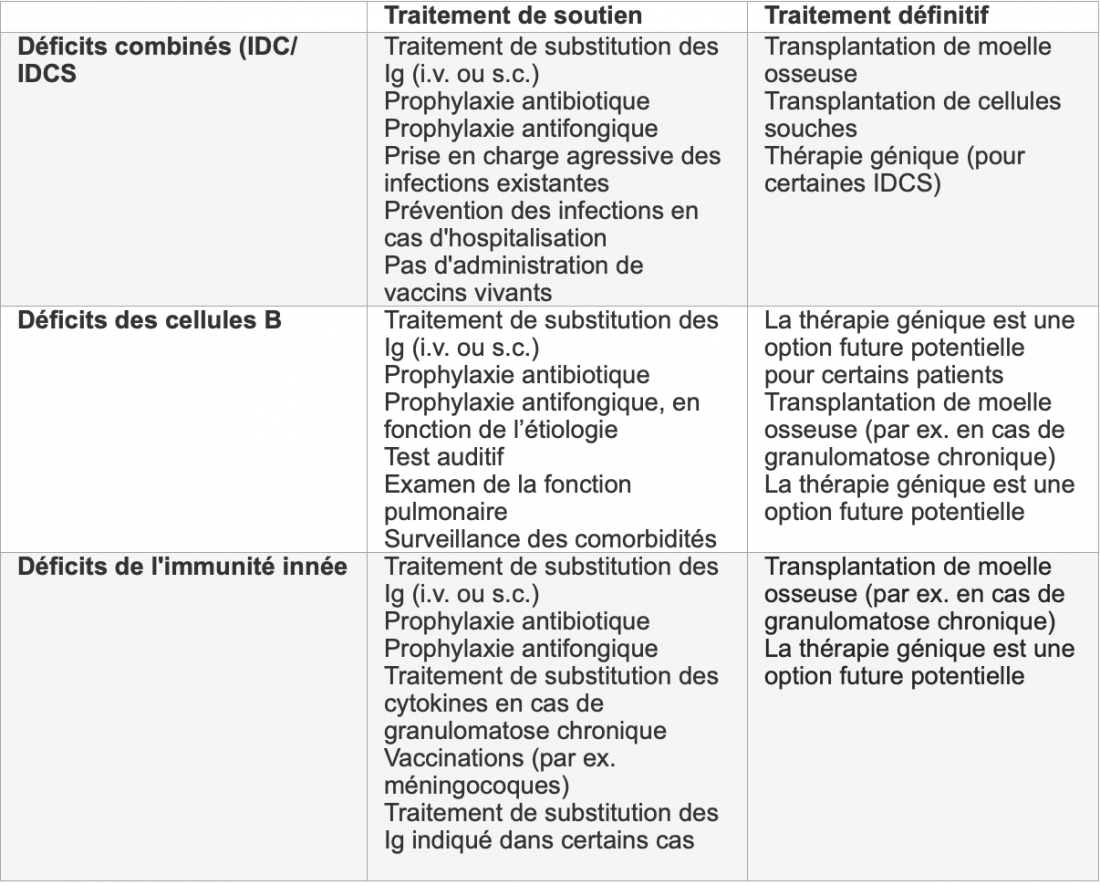
IDC: immunodéficience combinée, IDS: immunodéficience combinée sévère, i.v.: intraveineuse; s.c.: sous-cutanée, CGD: granulomatose chronique, Ig: immunoglobuline.
Traitement de substitution des Ig: des directives claires en cas de maladies liées à un déficit en anticorps
Sur la base des données publiées, le groupe de travail allemand des sociétés médicales scientifiques (AWMF) a rédigé une directive sur l’indication des traitements de substitution des Ig en cas de maladies liées à un déficit en anticorps (Tableau 2) [7]. Selon cette dernière, outre la sensibilité pathologique aux infections, une réponse vaccinale insuffisante représente un autre critère majeur pour l’indication d’un traitement de substitution des Ig. En cas d’urgence clinique, l’évaluation de la réponse vaccinale ne doit cependant pas retarder l’initiation d’un traitement de substitution des Ig.
L’efficacité du traitement de substitution des Ig a été démontrée dans diverses études en cas d’agammaglobulinémie ainsi qu’en cas d’hypogammaglobulinémie avec réponse vaccinale limitée [7]. Cela vaut également pour les patients atteints d’un déficit immunitaire commun variable (DICV), la maladie liée à un déficit en anticorps cliniquement pertinente la plus fréquente.
Tableau 2: Données sur le bénéfice d’un traitement de substitution des Ig dans le cadre de maladies liées à un déficit en anticorps spécifiques. Adapté d’après [7]
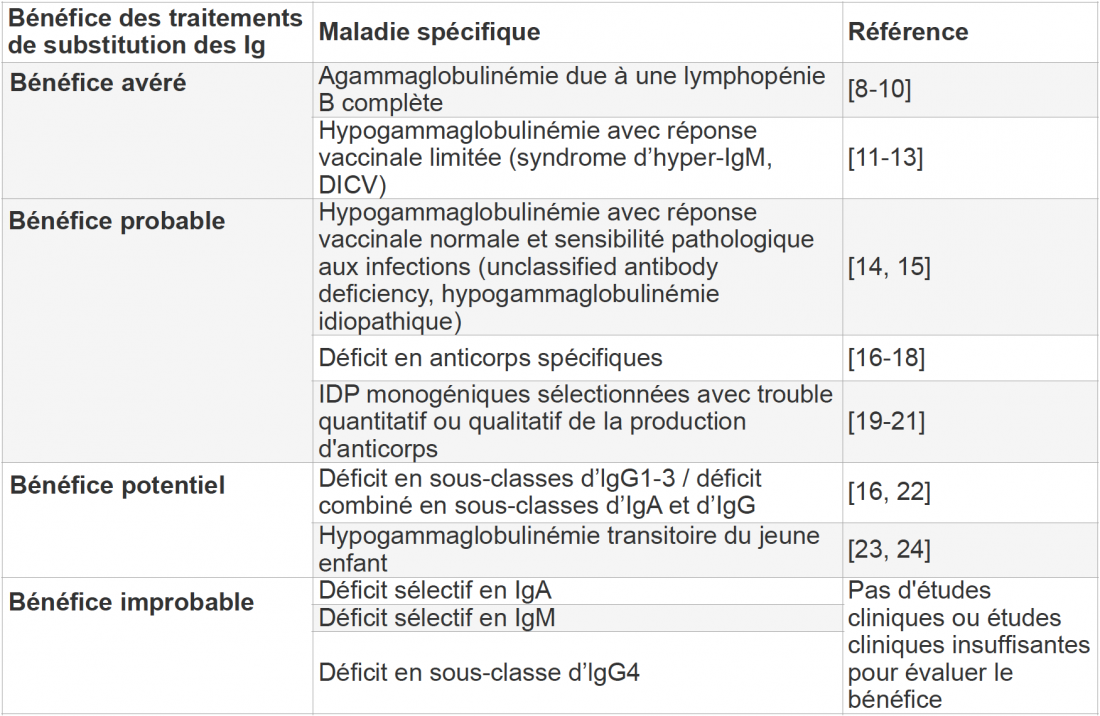
Ig: immunoglobulines, DICV: déficit immunitaire commun variable.
Approches immunomodulatrices en cas de manifestations non infectieuses
La prise en charge des manifestations auto-immunes et auto-inflammatoires constitue un défi de taille et est fonction des traitements déjà établis dans les différentes spécialités. Les patients atteints de DICV, en particulier, souffrent souvent de maladies auto-immunes, notamment de cytopénies auto-immunes, dont les récidives peuvent être bien traitées par glucocorticoïdes et rituximab [2]. Les traitements de substitution des Ig à dose élevée ont également montré une efficacité immunomodulatrice dans les thrombocytopénies auto-immunes et ils sont de plus en plus utilisés en présence de manifestations auto-immunes [25]. L’AWMF recommande un traitement de substitution des Ig chez les patients atteints de DICV, même en l’absence de sensibilité pathologique aux infections [7].
Transplantation de cellules souches et thérapie génique en cas de déficits combinés
Les maladies liées à un déficit en anticorps avec déficit immunitaire cellulaire supplémentaire peuvent nécessiter une transplantation de cellules souches hématopoïétiques, car le traitement par immunoglobulines et/ou substances immunomodulatrices à lui seul n’est souvent pas suffisant [2]. Les approches de thérapie génique, dans lesquelles des cellules souches autologues sont retransplantées après avoir remplacé le gène défectueux, ont déjà permis d’améliorer les chances de survie dans le cadre de certains déficits immunitaires combinés et font actuellement l’objet d’études quant à leur efficacité dans les déficits en cellules B et les déficits de l’immunité innée [6].
Les vaccins réduisent le risque d’infection
En cas de maladie liée à un déficit en anticorps avec limitation supplémentaire de l’immunité cellulaire, une antibioprophylaxie durable contre les agents pathogènes opportunistes peut être nécessaire. En outre, les patients atteints d’IDP devraient recevoir les vaccins recommandés, car même si leur efficacité n’est pas exactement prévisible chez ces patients, ils peuvent augmenter la protection contre les maladies infectieuses dangereuses. Les vaccins vivants sont toutefois contre-indiqués en cas d’IDP [3].
Conclusion
Un diagnostic précoce est essentiel pour pouvoir traiter une IDP de manière adéquate et prévenir les complications à long terme [2]. Un grand nombre d’études montrent que les traitements de substitution des Ig sont efficaces en cas de maladies liées à un déficit en anticorps, la forme la plus fréquente d’IDP [7]. Toutefois, une transplantation de cellules souches peut être nécessaire en cas de déficits combinés [2]. En tant que mesure de soutien, les vaccins peuvent augmenter la protection contre les maladies infectieuses dangereuses [3].
Bibliographie
1. Amaya-Uribe, L., et al., Primary immunodeficiency and autoimmunity: a comprehensive review. Journal of autoimmunity, 2019. 99: p. 52-72.
2. Göschl, L., et al., Diagnostik und Therapie bei primären Immundefekten/„inborn errors of immunity “. Wiener klinische Wochenschrift Education, 2019. 14(1): p. 65-79.
3. www.immunschwaeche-schweiz.ch. Letzter Abruf: Mai 2021.
4. Douglas Paes Barreto, I.C., et al., Immunological deficiencies: more frequent than they seem to be. 2020.
5. Gathmann, B., et al., Clinical picture and treatment of 2212 patients with common variable immunodeficiency. Journal of Allergy and Clinical Immunology, 2014. 134(1): p. 116-126. e11.
6. McCusker, C. and R. Warrington, Primary immunodeficiency. Allergy, Asthma & Clinical Immunology, 2011. 7(1): p. 1-8.
7. Hanitsch, L., et al., S3-Leitlinie: Therapie primärer Antikörpermangelerkrankungen. AWMF online, 2019.
8. Nolte, M., et al., Intravenous immunoglobulin therapy for antibody deficiency. Clinical and experimental immunology, 1979. 36(2): p. 237.
9. Quartier, P., et al., Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. The Journal of pediatrics, 1999. 134(5): p. 589-596.
10. Aghamohammadi, A., et al., Adverse reactions of prophylactic intravenous immunoglobulin infusions in Iranian patients with primary immunodeficiency. Annals of Allergy, Asthma & Immunology, 2004. 92(1): p. 60-64.
11. Skull, S. and A. Kemp, Treatment of hypogammaglobulinaemia with intravenous immunoglobulin, 1973-93. Archives of disease in childhood, 1996. 74(6): p. 527-530.
12. Winkelstein, J., The X-Linked Hyper-IgM Syndrome: Clinical and Immunologic Features of 79 Patients. The Pediatric Infectious Disease Journal, 2004. 23(10): p. 983.
13. Quartier, P., et al., Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clinical immunology, 2004. 110(1): p. 22-29.
14. Driessen, G.J., et al., Common variable immunodeficiency and idiopathic primary hypogammaglobulinemia: two different conditions within the same disease spectrum. haematologica, 2013. 98(10): p. 1617.
15. Janssen, L., et al., Mild hypogammaglobulinemia can be a serious condition. Frontiers in immunology, 2018. 9: p. 2384.
16. Bernatowska‐Matuszkiewicz, E., et al., Clinical efficacy of intravenous immunoglobulin in patients with severe inflammatory chest disease and IgG3 subclass deficiency. Clinical & Experimental Immunology, 1991. 85(2): p. 193-197.
17. Barlan, I.B., R.S. Geha, and L.C. Schneider, Therapy for patients with recurrent infections and low serum IgG3 levels. Journal of allergy and clinical immunology, 1993. 92(2): p. 353-355.
18. Abdou, N.I., et al., Efficacy of intravenous gammaglobulin for immunoglobulin G subclass and/or antibody deficiency in adults. International archives of allergy and immunology, 2009. 149(3): p. 267-274.
19. McKelvie, B., et al., Fatal pneumococcal meningitis in a 7-year-old girl with interleukin-1 receptor activated kinase deficiency (IRAK-4) despite prophylactic antibiotic and IgG responses to Streptococcus pneumoniae vaccines. Journal of clinical immunology, 2014. 34(3): p. 267-271.
20. Gobin, K., et al., IRAK4 deficiency in a patient with recurrent pneumococcal infections: case report and review of the literature. Frontiers in pediatrics, 2017. 5: p. 83.
21. Stentzel, S., et al., Reduced immunoglobulin (Ig) G response to Staphylococcus aureus in STAT3 hyper-IgE syndrome. Clinical Infectious Diseases, 2017. 64(9): p. 1279-1282.
22. Olinder-Nielsen, A.-M., et al., Immunoglobulin prophylaxis in 350 adults with IgG subclass deficiency and recurrent respiratory tract infections: a long-term follow-up. Scandinavian journal of infectious diseases, 2007. 39(1): p. 44-50.
23. Duse, M., et al., Transient hypogammaglobulinemia of infancy: intravenous immunoglobulin as first line therapy. International journal of immunopathology and pharmacology, 2010. 23(1): p. 349-353.
24. Memmedova, L., et al., Does intravenous immunoglobulin therapy prolong immunodeficiency in transient hypogammaglobulinemia of infancy? Pediatric reports, 2013. 5(3): p. 53-57.
25. Steiner, U. and M. Marconato, Patienten unter Therapie mit Immunglobulinen. Der informierte@ rzt, 2017: p. 34-35.
Ce texte a été rédigé avec le soutien financier de Takeda Pharma SA.
C-ANPROM/CH/CUVI/0013 09/2021
Article en ligne depuis le 27.01.2022