Les améliorations dans la gestion de la maladie augmentent la qualité et l’espérance de vie des patients atteints de mucoviscidose. Le dépistage néonatal, introduit en 2011, permet d’établir un diagnostic précoce. Cependant, elle doit également être considérée comme un diagnostic différentiel possible à l’âge adulte.
La fibrose kystique (mucoviscidose) est la maladie métabolique congénitale la plus fréquente en Suisse, dont l’issue est fatale. En tant que maladie systémique, la mucoviscidose affecte principalement le fonctionnement des glandes exocrines des voies respiratoires et du tube digestif. C’est notamment l’évolution pulmonaire avec destruction continue et irréversible de l’organe qui conditionne la réduction de l’espérance de vie. En raison de l’amélioration des mesures médicales, les personnes concernées atteignent désormais presque toutes l’âge adulte, mais elles constituent toujours un groupe relativement nouveau dans la pratique de la médecine générale. Nouveauté prometteuse, un traitement causal est disponible depuis début 2014 pour un très petit nombre de patients.
Tableau clinique
Dès 1936, le pédiatre zurichois Guido Fanconi a décrit la mucoviscidose, alors appelée autrement, comme une maladie mortelle chez les jeunes enfants, ce qui explique pourquoi les collègues médecins généralistes n’y ont pas eu accès pendant longtemps.
La mucoviscidose est la maladie héréditaire congénitale chronique la plus fréquente, dont la prévalence est d’environ 1 sur 2500. Environ une personne sur 25 en Europe centrale est porteuse d’une mutation saine. On compte environ 70 000 malades dans le monde, et on estime tout de même à 900 le nombre de personnes atteintes en Suisse. Environ la moitié des patients ont plus de 18 ans. La transmission est autosomique récessive, ce qui signifie que statistiquement, sur quatre enfants issus d’une relation entre deux porteurs de mutation sains, un enfant est atteint de mucoviscidose, deux enfants sont également porteurs de mutation sains comme leurs parents et un enfant n’est ni porteur de mutation ni malade. En 1989, le gène sous-jacent a été identifié sur le chromosome 7. La cause est une mutation dans le gène CFTR (“cystic fibrosis transmembrane conductance regulator”), qui code pour le canal chlorure dans la membrane cellulaire [1,2].
Clinique
Dans le cas de la maladie systémique, différents organes sont touchés, mais en règle générale, la maladie pulmonaire chronique entraîne une morbidité accrue et une espérance de vie réduite. Grâce à l’amélioration des possibilités médicales, l’espérance de vie n’a cessé d’augmenter ces dernières années. L’espérance de vie moyenne est actuellement de plus de 40 ans en Europe et continuera d’augmenter, en particulier pour la génération détectée par le dépistage néonatal en Suisse depuis 2011. Malgré cela, l’évolution de la maladie est très inégale et certains malades décèdent encore d’une insuffisance pulmonaire à l’âge adulte. Toutes les personnes atteintes ont en commun des infections bactériennes chroniques des voies respiratoires qui entraînent une destruction irréversible des poumons. Les patients souffrent de toux, d’expectorations et d’une diminution progressive de leur capacité physique. En outre, l’insuffisance pancréatique exocrine, qui touche environ 85% des patients, entraîne une diarrhée chronique en cas de maldigestion. En conséquence de la malnutrition, mais aussi d’un travail respiratoire accru, il en résulte un trouble de la croissance. Environ 10% d’entre eux présentent un iléus méconial dès la naissance, mais les personnes plus âgées peuvent également souffrir de syndromes d’obstruction intestinale récurrents. Malgré un traitement symptomatique optimal, l’évolution de la maladie peut en outre conduire à une cirrhose du foie ou à une insuffisance pancréatique endocrine avec développement d’un diabète sucré. Les hommes atteints de mucoviscidose, en particulier, sont souvent touchés par l’infertilité [1,2].
Physiopathologie
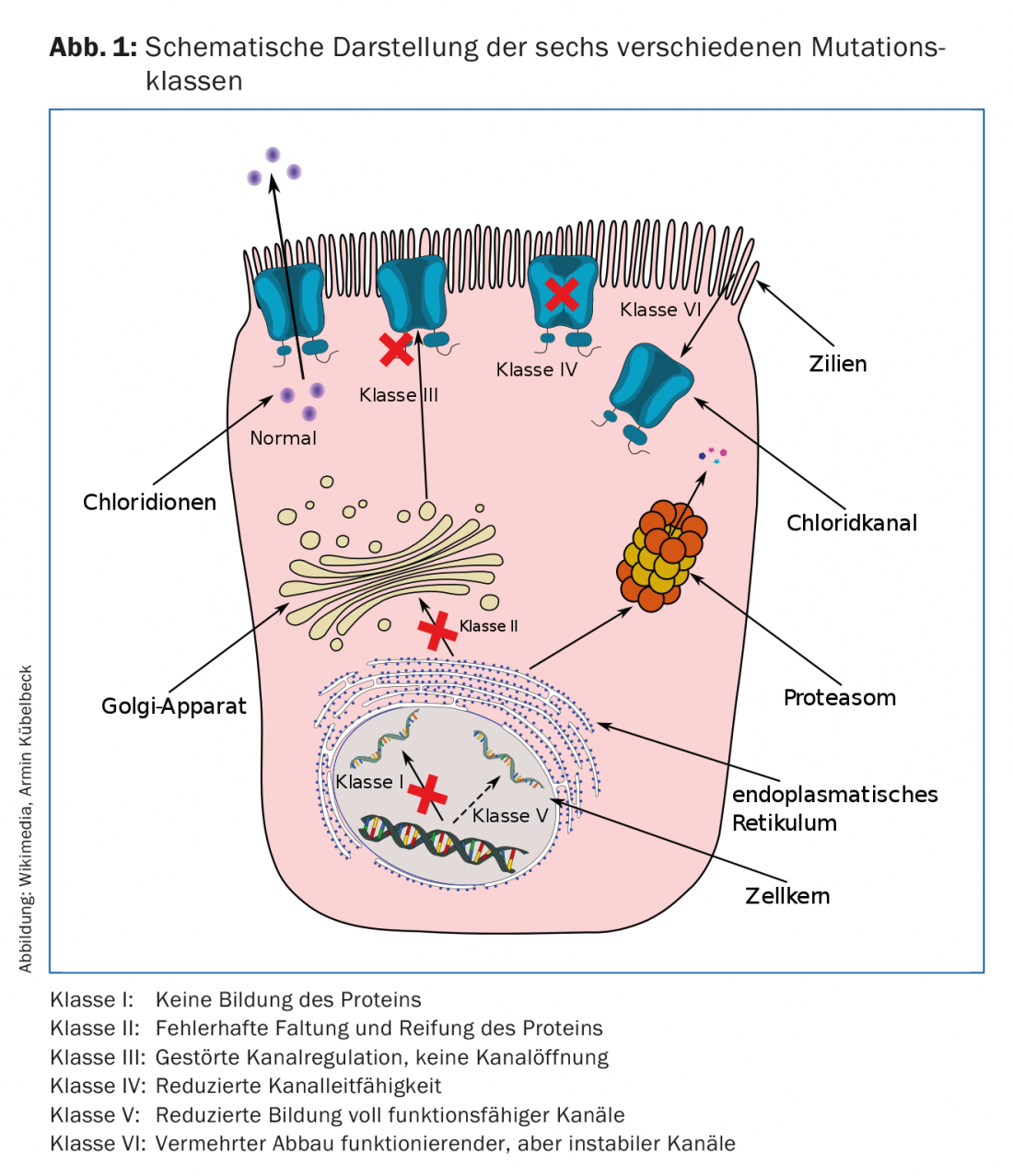
Le gène CFTR, situé sur le chromosome 7, code pour le canal chlorure dans la membrane cellulaire. Les mutations génétiques, dont plus de 2000 sont désormais connues mais dont moins de 200 sont clairement responsables de la maladie, entraînent un dysfonctionnement ou une absence totale du canal chlorure dans les cellules épithéliales. Les mutations du gène CFTR sont divisées en six classes (Fig. 1), qui se distinguent par leur pathomécanisme. Certaines mutations entraînent par exemple une absence quasi-totale de synthèse de la protéine CFTR. La mutation 3905insT, qui se produit chez les Suisses ou les personnes d’origine suisse (par exemple, les Amish en Amérique du Nord), appartient à cette classe I et constitue la deuxième mutation la plus fréquente en Suisse. Dans d’autres mutations, l’intégration de la protéine dans la membrane cellulaire est empêchée ou le canal ionique de la protéine est bloqué ou ne présente qu’une conductivité limitée. La mutation la plus fréquente en Europe, mais aussi en Suisse (86% hétérozygote, 47% homozygote) est classée en classe II et concerne une délétion en position 508 (F508del). Les protéines produites sont ici mal repliées et dégradées avant même d’entamer une éventuelle fonction. La défaillance du canal chlorure entraîne une perturbation du transport transépithélial dans tous les organes où les cellules épithéliales expriment CFTR. Si l’on prend l’exemple de l’épithélium des voies respiratoires, il en résulte une cascade d’inflammations. L’augmentation de la viscosité du mucus entraîne une perturbation de la clairance mucociliaire, qui s’accompagne à son tour d’une colonisation bactérienne précoce et, par la suite, chronique. La réponse immunitaire de l’organisme provoque un recrutement excessif de granulocytes neutrophiles, une sécrétion inadéquate d’élastase neutrophile et, finalement, une destruction des tissus. Outre les deux mutations classiques citées à titre d’exemple (F508del, 3905insT), qui sont responsables d’une évolution grave de la maladie, on connaît désormais des mutations qui s’accompagnent d’une clinique plutôt bénigne – alors que pour d’autres mutations encore, on ne sait pas si elles sont vraiment à l’origine de la mucoviscidose. La base de données www.cftr2.org tente de fournir une vue d’ensemble constamment mise à jour des mutations connues déclenchant la mucoviscidose et de leurs phénotypes probables [1,3,4].
Diagnostic
Avant 2011, environ 10% des enfants malades présentaient un iléus méconial, mais la grande majorité n’était identifiée qu’au cours de l’évolution, lorsque des symptômes suspects de mucoviscidose apparaissaient. Le test de la sueur, c’est-à-dire la détermination quantitative de la concentration de chlorure dans la sueur après ionophorèse à la pilocarpine, est considéré comme le gold standard pour le diagnostic. En outre, la mesure de la conductivité de la sueur (conductivité) est souvent utilisée comme test de dépistage, car ce test fonctionne rapidement et de manière fiable avec une quantité de sueur nettement plus faible. Macroduct® et Nanoduct® sont des systèmes de test disponibles dans le commerce. Pour l’assurance qualité, la réalisation de tests de soudage devrait être réservée à des centres appropriés.
En 2011, la mucoviscidose a été incluse dans le programme de dépistage néonatal en Suisse. Le trypsinogène immunoréactif (IRT) est mesuré dans le sang du talon. En cas de dépassement du seuil, les nouveau-nés sont dirigés vers un centre de mucoviscidose pédiatrique. Le diagnostic y est confirmé ou exclu par un test à la sueur et, le cas échéant, par des examens complémentaires (détermination de l’élastase pancréatique, analyse génétique).
Jusqu’en 2016, le dépistage néonatal a permis de diagnostiquer 143 enfants atteints de mucoviscidose. Sur un total estimé de 900 patients atteints de mucoviscidose, le dépistage néonatal a déjà permis de détecter environ 15% des malades. Cette valeur ne cessera d’augmenter.
Toutefois, il convient de noter, en particulier pour le médecin généraliste, qu’en raison des différents phénotypes, il peut y avoir, outre les cas diagnostiqués dans l’enfance, ceux dont l’évolution est légère ou dont la fonction CFTR résiduelle est mesurable. Certains individus peuvent donc ne se manifester qu’à l’âge adulte avec une symptomatologie atypique comme une pancréatite, une sinusite, des polypes nasaux, des bronchectasies diffuses et/ou un désir d’enfant inassouvi. Par conséquent, le médecin généraliste devrait également envisager la mucoviscidose comme un diagnostic différentiel possible en cas de symptômes correspondants [1,5–7].
Thérapie et soins
Selon les directives internationales, les malades doivent être rattachés à des centres spécialisés. Les patients sont vus tous les trois mois. En fonction de l’évolution de la maladie, le traitement doit être revu et adapté. Outre l’anamnèse et l’examen clinique, les examens de routine comprennent des mesures de la fonction pulmonaire (selon l’infrastructure, bodyplethysmographie, spirométrie, N2-multiple-breath-washout) et des tests microbiologiques (expectoration ou prélèvement de gorge), complétés à intervalles plus longs par des imageries des poumons et de l’abdomen, des analyses de sang, des charges en glucose et des mesures de la densité osseuse. L’objectif est de détecter rapidement les changements afin de les contrer et de prévenir toute nouvelle détérioration.
Le traitement de base est lourd et comprend la clairance intensive des voies respiratoires par inhalation et physiothérapie respiratoire spéciale (solution saline hypertonique, rhDNAse si nécessaire, “technique spéciale de clairance des voies respiratoires”), la thérapie par enzymes pancréatiques (lipase) et nutritionnelle (substitution des vitamines liposolubles, régime hypercalorique) ainsi qu’un traitement antimicrobien agressif (application par inhalation et/ou par voie systémique).
En fonction de l’âge du patient et des circonstances qui l’accompagnent, des conseils continus et ciblés sont également nécessaires pour relever au mieux les défis de la maladie et son impact sur la vie. Outre les contenus évidents liés à la maladie et au traitement, les patients (et, en fonction de leur âge, leurs tuteurs) ont également besoin d’un soutien pour aborder les questions sociales, psychologiques, financières, d’assurance (invalidité et/ou maladie), d’organisation de la fréquentation de la crèche, du jardin d’enfants et de l’école, de la formation et du travail, des voyages, du nouveau désir d’enfant des parents ou du propre planning familial des adultes malades, etc.
Il faut en outre tenir compte du fait que les personnes concernées sont heureusement de plus en plus âgées. Soudain, les patients, mais aussi les équipes soignantes, sont exposés à des problèmes internes qui surviennent indépendamment de la mucoviscidose, mais qui contribuent à en influencer l’évolution. Pour répondre à ces exigences, une approche thérapeutique multidisciplinaire est nécessaire, ce que seul un centre CF peut faire [8,9].
Perspectives
En 2014, Swissmedic a autorisé le médicament Ivacaftor (Kalydeco®) pour les patients atteints de la mutation de classe III G551D. Pour la première fois, un traitement causal a pu être appliqué aux quelques patients atteints de cette mutation (environ 4 à 5% de tous les patients dans le monde), avec une augmentation significative de la fonction des canaux chlorure, mesurable par une amélioration significative du test de la sueur et de la fonction pulmonaire. D’autres modulateurs CFTR sont actuellement à l’étude. Il reste à espérer qu’à l’avenir, un traitement causal spécifique à la mutation sera disponible pour un plus grand nombre de patients [10].
Messages Take-Home
- Grâce aux améliorations apportées au traitement symptomatique et à la gestion de la maladie, la qualité et l’espérance de vie ne cessent d’augmenter.
- Aujourd’hui déjà, la moitié des patients suisses atteints de mucoviscidose sont des adultes.
- Le dépistage néonatal de la mucoviscidose, introduit en 2011, permet de poser un diagnostic précoce. Cela permet de commencer un traitement de base avant même l’apparition des premiers changements liés à la maladie.
- Des évolutions oligosymptomatiques et atypiques sont possibles. Par conséquent, la mucoviscidose doit également être considérée comme un diagnostic différentiel possible à l’âge adulte.
- L’évaluation et le suivi doivent toujours être effectués en collaboration avec un centre de mucoviscidose.
Littérature :
- Elborn JS : Mucoviscidose. Lancet 2016 Nov 19 ; 388(10059) : 2519-2531.
- Zolin A, et al. : ECFSPR Rapport annuel 2014. 2016.
- Hergersberg M, et al : Une nouvelle mutation, 3905insT, représente 4.8% des 1173 chromosomes CF en Suisse et provoque un phénotype sévère. Hum Genet 1997 Aug ; 100(2) : 220-223.
- CFTR2. www.cftr2.org
- Torresani T, et al : Newborn screening for cystic fibrosis in Switzerland–consequences after analysis of a 4 months pilot study. J Cyst Fibros 2013 ; 12(6) : 667-674.
- Dépistage néonatal en Suisse. www.neoscreening.ch
- Barben J, et al : Dépistage néonatal de la mucoviscidose – une histoire à succès. Forum Med Suisse 2013 ; 13(49) : 1010-1012.
- Smyth AR, et al : Standards de soins de l’European Cystic Fibrosis Society : lignes directrices de bonne pratique. J Cyst Fibros 2014 May ; 13(Suppl 1) : S23-42.
- Elborn JS, et al : Rapport du groupe de travail de l’European Respiratory Society/European Cystic Fibrosis Society sur les soins des adultes atteints de mucoviscidose. Eur Respir J 2016 Feb ; 47(2) : 420-428.
- Ramsey BW, et al : A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011 Nov 3 ; 365(18) : 1663-1672.
PRATIQUE DU MÉDECIN DE FAMILLE 2017 ; 12(11) : 31-33









