La valve aortique bicuspide (BAK) est une cardiopathie congénitale fréquente. Outre la dysfonction valvulaire, elle se traduit parfois par une dilatation de la racine de l’aorte ou de l’aorte ascendante. Cette mise à jour pratique fournit des informations sur le diagnostic, la coarctation de l’aorte en tant que vitium le plus fréquent et la nécessité d’un dépistage familial.
La valve aortique bicuspide (BAK) est une cardiopathie congénitale fréquente qui touche environ 1 à 2 % de la population. Elle a été décrite pour la première fois par Léonard de Vinci il y a plus de 400 ans. La malformation cardiovasculaire associée la plus fréquente est la coarctation de l’isthme aortique (AIS), bien que des formes plus rares d’obstruction congénitale de la voie de sortie du ventricule gauche puissent également être associées. Environ 40 à 50 % des patients ayant une valve aortique bicuspide présentent une dilatation de la racine aortique ou de l’aorte ascendante [1]. L’ampleur de la dilatation n’est souvent pas corrélée à l’ampleur du dysfonctionnement valvulaire. L’aortopathie sous-jacente ainsi que la géométrie de la valve aortique bicuspide et les caractéristiques du flux qui en découlent ont une influence importante sur la localisation et l’étendue de la dilatation. Les dissections aortiques sont relativement rares, malgré la dilatation aortique fréquemment observée. La complication à long terme la plus fréquente de l’AFC est le développement d’une insuffisance aortique progressive et/ou d’une sténose aortique. Par rapport aux personnes en bonne santé cardiaque, le risque d’endocardite est nettement plus élevé chez les patients souffrant d’AC (13,9 pour 10 000 années-patients, soit un risque relatif multiplié par 11,4 par rapport à la population normale) [2].
Une bonne éducation des patients et une bonne hygiène dentaire sont donc importantes, tandis que la protection antibiotique lors d’interventions dentaires (prophylaxie de l’endocardite) n’est plus recommandée que pour les patients ayant subi un remplacement valvulaire prothétique ou une endocardite. Il arrive qu’une opération soit nécessaire dès l’enfance. Dans le cas des valves aortiques bicuspides, la dégénérescence de la valve, qui nécessite un remplacement valvulaire aortique, survient en moyenne une à deux décennies plus tôt que chez les personnes dont les valves aortiques tricuspides sont dégénérées. L’éventail des âges au moment où l’indication d’une intervention est posée est toutefois large. La reconstruction ou le remplacement de la valve aortique ne permet pas de “guérir” la valve aortique bicuspide et les patients concernés doivent faire l’objet d’un suivi cardiaque à vie. Comme les valves aortiques bicuspides sont fréquentes dans les familles, il est recommandé de procéder à un dépistage échocardiographique de tous les membres de la famille au premier degré.
Diagnostic
Les valves aortiques bicuspides dysplasiques sont diagnostiquées in utero, peu après la naissance ou pendant l’enfance, selon la gravité du dysfonctionnement de la valve (généralement une sténose). En cas de sténose aortique critique, une instabilité hémodynamique apparaît souvent immédiatement après la naissance, tandis qu’en cas de dysfonctionnement valvulaire moins prononcé, c’est généralement le souffle cardiaque typique qui conduit au diagnostic. Chez les patients présentant une valve aortique bicuspide avec une fonction valvulaire normale, un clic systolique précoce à l’auscultation cardiaque est souvent le seul résultat anormal à l’examen clinique. Il n’est donc pas rare que la valve aortique bicuspide soit une découverte fortuite à l’échocardiographie. Chez les patients asymptomatiques présentant un dysfonctionnement de la valve aortique bicuspide, c’est souvent l’auscultation anormale ou la clinique qui conduit au diagnostic.
En cas de coexistence d’une coarctation de l’aorte (environ 6% des patients présentant une valve aortique bicuspide [3]), les signes cliniques de la coarctation de l’aorte (hypertension artérielle de la moitié supérieure du corps et différence de pression artérielle entre les bras et les jambes), l’affaiblissement des pouls fémoraux ou, dans les formes prononcées, une insuffisance cardiaque sévère, conduisent au diagnostic. Selon le collectif, une valve aortique bicuspide est également diagnostiquée chez les patients présentant une coarctation de l’aorte dans environ 50% des cas [4]. Les adolescents et les adultes peuvent également présenter un syndrome de coarctation – l’association d’une coarctation de l’isthme aortique, d’une valve aortique bicuspide et d’un anévrisme de l’aorte ascendante.
D’autres malformations cardiaques congénitales sont également associées à une valve aortique bicuspide et à une coarctation de l’isthme aortique. Il convient de mentionner le complexe de Shone, dans lequel, outre l’obstruction de la voie de sortie du ventricule gauche à différents niveaux (sténose sous-aortique, valve aortique bicuspide, coarctation de l’isthme aortique), l’afflux ventriculaire gauche est également anormal avec une sténose mitrale membraneuse supravalvulaire et une valve mitrale dysplasique, souvent sous la forme d’une “parachute mitral valve”.
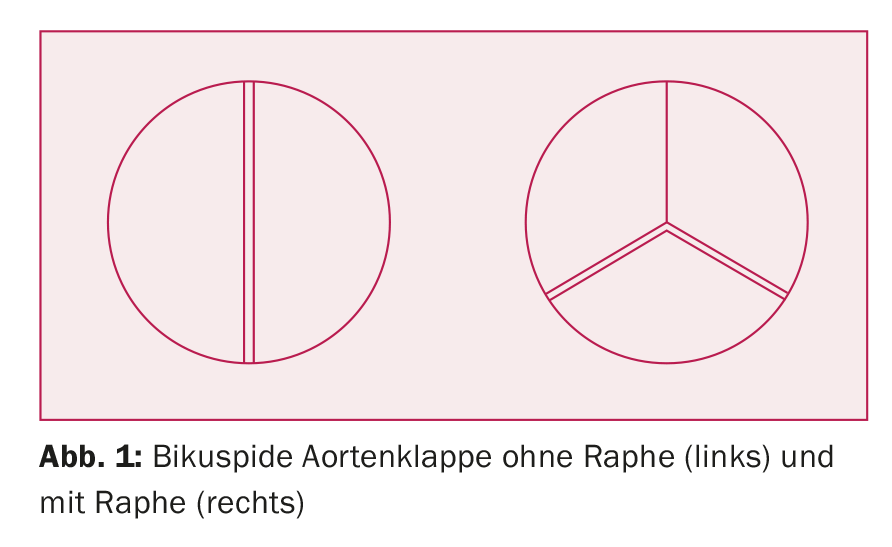
La BAK est généralement détectée par échocardiographie et parfois découverte par hasard lors d’un examen IRM cardiaque. Le diagnostic par tomodensitométrie (TDM) est plus difficile, car pour la plupart des indications, l’acquisition des images se fait en (fin)diastole, la valve aortique étant fermée. Si la cuspidie ne peut pas être clairement identifiée par voie transthoracique en raison de la mauvaise qualité des fenêtres d’échographie, le diagnostic peut être établi par échocardiographie transoesophagienne. Lorsque les valves sont fortement calcifiées, il peut parfois être difficile de faire la distinction entre une valve aortique bicuspide et tricuspide. Il existe différentes variantes de valves aortiques bicuspides : On distingue les valves aortiques bicuspides “vraies”, dans lesquelles la valve est constituée de deux poches de taille égale (environ 20% de toutes les valves bicuspides), et les valves aortiques tricuspides, dans lesquelles une (ou plus rarement deux) poche(s) est/sont fusionnée(s) au niveau des commissures pour former un raphé. (Fig. 1). La fusion des poches coronaires droite et gauche est la plus fréquente. Si toutes les poches de deux commissures sont fusionnées ou si une seule poche est formée, la valve est dite monocuspide.
En cas de suspicion de coexistence d’une coarctation de l’aorte, il est indispensable de confirmer le diagnostic par angio-IRM ou angio-TDM. Il convient de noter que les patients dont la circulation collatérale est bien établie sont peu symptomatiques et qu’un profil de flux abdominal anormal dans l’aorte abdominale est le seul signe échocardiographique de la présence d’une coarctation de l’aorte. Il est important d’envisager un diagnostic différentiel avec la présence d’une coarctation de l’aorte chez les enfants ou les jeunes adultes souffrant d’hypertension artérielle systémique, car il s’agit de l’une des rares causes d’hypertension artérielle secondaire pouvant être traitée. Le diagnostic peut presque toujours être établi par une simple mesure de la pression artérielle au niveau des bras et des jambes avec mise en évidence d’un gradient de pression artérielle.
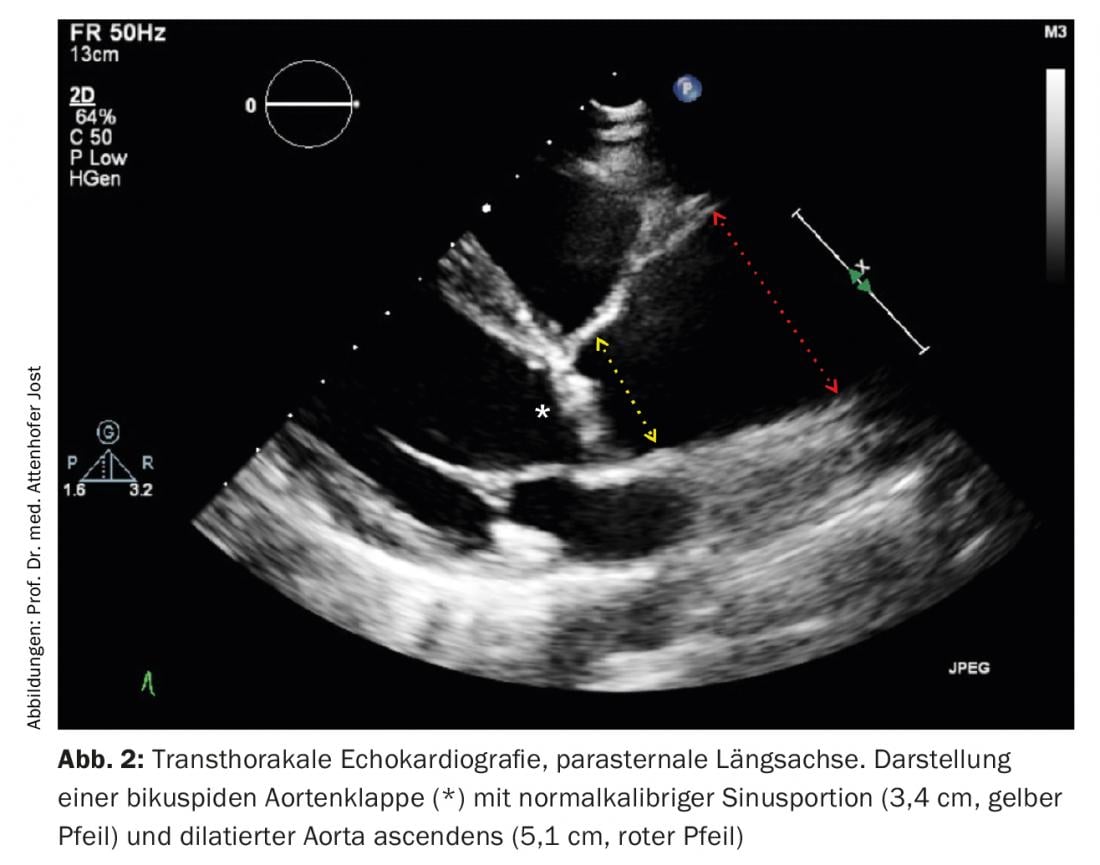
Selon l’ampleur et le type de dysfonctionnement valvulaire présent (sténose aortique et/ou insuffisance aortique), les patients asymptomatiques doivent faire l’objet d’un suivi cardiologique et échocardiographique régulier afin de détecter à temps une progression. C’est particulièrement vrai pour les patients atteints d’insuffisance aortique pré-dominante, dont les symptômes apparaissent souvent tard dans l’évolution de la maladie, lorsque le myocarde du ventricule gauche est déjà irréversiblement endommagé. De même, la dilatation de l’aorte nécessite des contrôles réguliers afin de permettre, en cas de progression, un remplacement aortique en temps utile pour des raisons pronostiques. La figure 2 montre un exemple échocardiographique typique d’un patient présentant une valve aortique bicuspide et un rétrécissement aortique ainsi qu’une dilatation de l’aorte ascendante.
Anévrisme de l’aorte en cas d’alcoolémie
La dilatation de la racine de l’aorte et de l’aorte ascendante, généralement indépendante de la gravité du dysfonctionnement de la valve aortique, est plus fréquente chez les patients souffrant d’ACA. Selon des données récentes sur les patients, le risque de dissection ou de rupture de l’aorte ne semble pas fortement augmenté par rapport aux patients ayant une valve aortique tricuspide [5]. Les principaux facteurs de risque de complications aortiques sont l’abus de nicotine, une hypertension artérielle systémique insuffisamment traitée, la présence simultanée d’une coarctation de l’aorte ou d’antécédents familiaux de dissection. Les valves aortiques bicuspides étant fréquentes, elles sont parfois présentes de manière coïncidente chez les patients atteints du syndrome de Marfan ou d’autres aortopathies héréditaires. En cas d’indices cliniques d’une maladie du tissu conjonctif, il vaut donc la peine de procéder à un examen génétique plus approfondi dans des cas sélectionnés.
Coarctation de l’aorte (AIS)
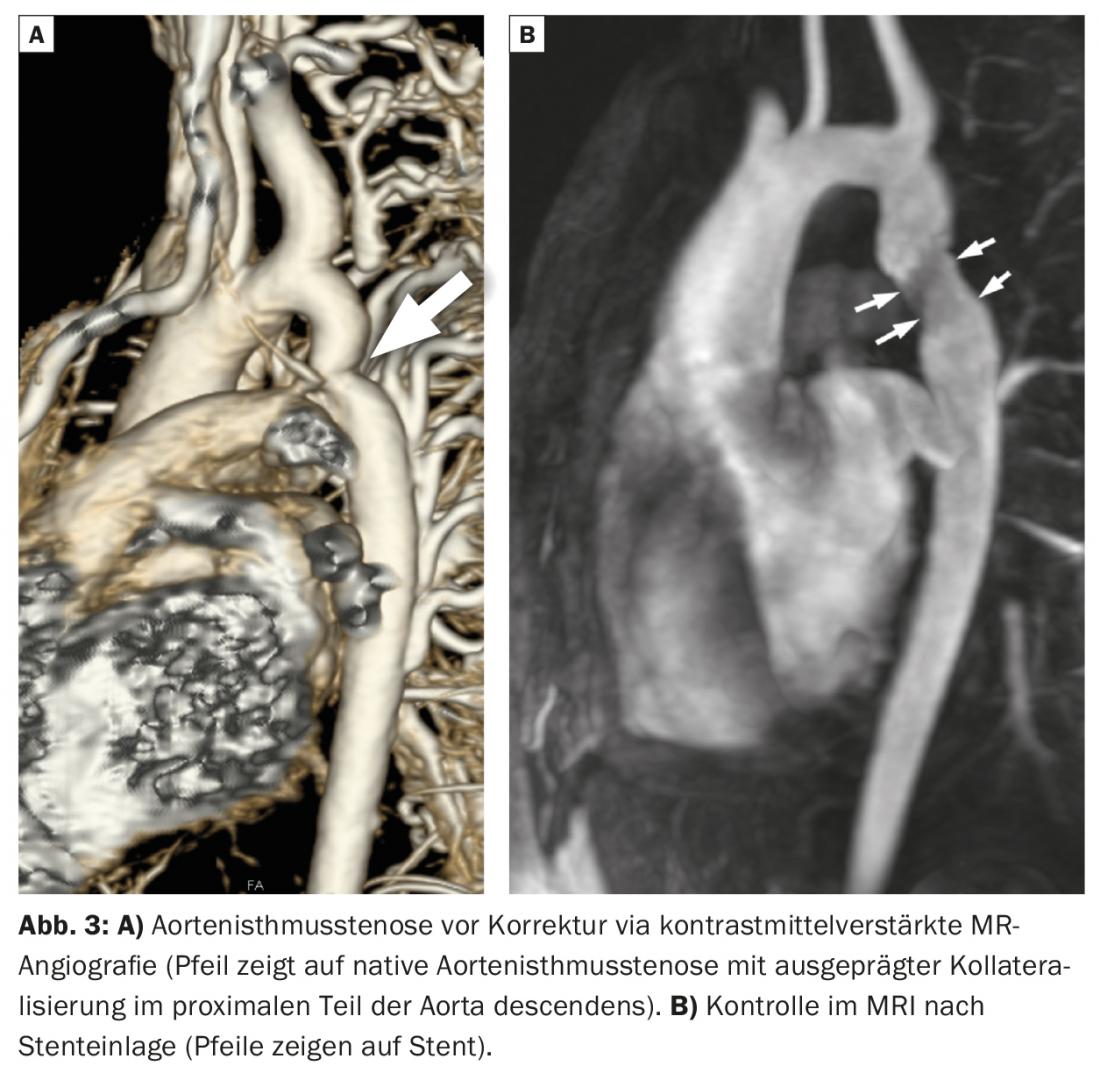
L’AIS est un rétrécissement dans la région de l’aorte descendante proximale. Les symptômes typiques sont une différence de pression artérielle avec des valeurs de pression artérielle plus élevées aux extrémités supérieures et inférieures, ainsi qu’une hypertension artérielle. La coarctation de l’aorte doit être traitée par résection (avec anastomose de bout en bout avec/sans dilatation du patch), par remplacement du segment aortique malade par un greffon ou par intervention avec dilatation par ballonnet et pose d’un stent. Un exemple de patient présentant un AIS avant et après la pose d’un stent est illustré à la figure 3. Les patients atteints d’AIS présentent un risque élevé de complications cardiovasculaires à long terme. Les complications les plus fréquentes sont l’hypertension artérielle systémique (même si le résultat de l’opération est bon !), la coarctation de l’isthme aortique avec nécessité de réintervention, la formation de pseudo-anévrismes au niveau du site de réparation chirurgicale ou interventionnelle et un risque accru d’anévrisme cérébral. Tous les patients atteints de coarctation de l’aorte doivent donc être pris en charge dans des centres spécialisés.
Le sport chez BAK et AIS
Il n’y a pas de restrictions concernant les activités sportives compétitives pour les athlètes ayant un taux d’alcoolémie et des altérations valvulaires légères au maximum ainsi qu’une aorte de calibre normal. Pour les sportifs ayant une BCA et une dilatation de l’aorte (portion sinusale/aorte ascendante), les sports à potentiel de collision (sports de contact) et les sports de force purs sont déconseillés, selon l’importance de la dilatation aortique. Lors d’un entraînement de musculation accompagné d’une charge isométrique maximale, la pression artérielle systolique peut augmenter jusqu’à 300-500 mmHg ! En cas de doute, il est essentiel de demander conseil à un spécialiste.
Suivi des patients avec BAC
Les patients souffrant d’une BCA ont parfois besoin d’un suivi auprès d’un cardiologue expérimenté dans ce domaine. Les intervalles de contrôle dépendent du degré de dysfonctionnement de la valve aortique et de l’étendue de l’aortopathie et doivent être déterminés individuellement.
Chez les femmes présentant une AFC, une échocardiographie fœtale est recommandée entre la 18e et la 20e semaine de grossesse, en raison du risque héréditaire accru de vitites dans la voie d’éjection du ventricule gauche (dans les cas extrêmes, syndrome du cœur gauche hypoplasique). En présence d’un valvulus aortique plus que léger ou d’une aorte dilatée, une consultation multidisciplinaire complète sur les risques de la grossesse est recommandée dans des centres spécialisés.
Dépistage familial, génétique et grossesse
Les valves aortiques bicuspides sont fréquentes dans les familles. Les hommes sont plus souvent touchés. On pense qu’il s’agit d’une transmission héréditaire dominante avec une pénétrance incomplète. Ainsi, chez les patients atteints, environ 9 à 11% des membres de la famille du premier degré présentent également une BAK ou, plus rarement, une dilatation aortique avec une valve tricuspide [6]. En outre, d’autres formes d’obstruction de la voie de sortie du ventricule gauche sont plus fréquentes dans les familles avec un AC, comme la coarctation de l’aorte et, comme “variante extrême”, le syndrome du cœur gauche hypoplasique.
Les antécédents familiaux doivent donc être recueillis pour chaque patient souffrant d’une maladie cardiaque. Il est recommandé de procéder au dépistage des membres de la famille au premier degré. Cependant, le dépistage génétique n’est pas encore une pratique courante.
Environ 20% des patients atteints de vities congénitales présentent un syndrome (par exemple, le syndrome de Turner, le syndrome de Down, le syndrome de DiGeorge). Les indices sont les problèmes intellectuels, les problèmes d’apprentissage, l’autisme, les traits du visage dysmorphiques, la petite taille ou les problèmes d’audition et de vision. Le syndrome le plus courant associé à un AAC (et à un AIS) est le syndrome de Turner (généralement monosomie X0). Il faut toujours y penser, car dans ce cas, un conseil génétique concernant la planification de la descendance serait recommandé.
Si le père a une sténose aortique, le risque de cardiopathie congénitale chez la progéniture est de 3 à 4 %, et de 8 à 18 % en cas de maladie de la mère. En ce qui concerne la coarctation de l’aorte, le risque pour la descendance est de 2-3% si le père est atteint et de 4-6,5% si la mère est atteinte.
Une sténose aortique importante est mal tolérée en cas de grossesse, c’est pourquoi le remplacement valvulaire doit être effectué avant. Toutefois, la question se pose alors de savoir s’il faut choisir une valve biologique, une valve mécanique ou une opération de Ross ; dans cette dernière option, la valve aortique est remplacée par la propre valve pulmonaire avec un remplacement artificiel de la valve en position pulmonaire. Chacune de ces variantes peut entraîner des complications à long terme, telles qu’une dégénérescence précoce (valve biologique), une anticoagulation (valve mécanique) ou des interventions répétitives (opération de Ross).
Risque d’endocardite
Comme pour toutes les vities congénitales, le risque d’endocardite est élevé dans le cas de la BAK et se produit chez jusqu’à 15% des patients. 50% des cas d’insuffisance aortique sévère chez les BAC sont dus à une endocardite parfois non diagnostiquée. Les recommandations habituelles, telles que le prélèvement d’hémocultures avant l’administration d’antibiotiques, s’appliquent pour l’établissement du diagnostic. On ne saurait trop insister sur l’importance de sensibiliser les patients atteints d’ACB et leurs médecins de premier recours aux symptômes typiques de l’endocardite et à la nécessité de procéder à un examen correct en cas d’apparition de ces symptômes.
Opérations et interventions
En cas de sténose ou d’insuffisance aortique, les critères de remplacement ou d’intervention sont les mêmes que pour la valve aortique tricuspide. Cependant, ces changements surviennent en moyenne une à deux décades plus tôt dans la vie. L’implantation percutanée de valves en présence d’une CBA n’est possible qu’en cas de sténose aortique et dans des cas soigneusement sélectionnés. La chirurgie reste ici le traitement de choix. Si un anévrisme de l’aorte ascendante est également présent, un remplacement de celle-ci est indiqué. Lorsque la CBA n’est pas calcifiée, une reconstruction, c’est-à-dire une intervention visant à préserver la valve, peut être envisagée dans des cas sélectionnés.
Les risques liés à la chirurgie de l’aorte ascendante et de la valve aortique dépendent des comorbidités du patient, du type de chirurgie et de l’expérience du centre chirurgical et du chirurgien. Chez les patients sans facteurs de risque supplémentaires, la mortalité entre des mains expertes <est de 1%, alors qu’une opération en urgence dans le cadre d’une dissection aiguë est associée à une mortalité d’environ 25% [7].
L’indication et le type de réintervention chez les patients souffrant de coarctation de l’aorte doivent être déterminés dans des centres expérimentés. En cas de coarctation de l’aorte, il est préférable de recourir à un traitement interventionnel par stenting, en raison du risque plus élevé de réintervention. Il faut toutefois évaluer l’anatomie individuelle du patient et planifier l’intervention en conséquence (par ex. en cas d’hypoplasie supplémentaire de l’arc aortique ou de sténoses au niveau des sorties des vaisseaux supra-aortiques).
Messages Take-Home
- La valve aortique bicuspide (VAB) est une malformation cardiaque fréquente qui touche environ 1 à 2 % de la population. En plus d’un dysfonctionnement prématuré de la valve aortique bicuspide, une dilatation de la racine aortique ou de l’aorte ascendante est souvent observée.
- Le vitium associé le plus fréquent est la coarctation de l’aorte.
- Un examen échocardiographique des membres de la famille au premier degré est recommandé.
- Les patients atteints d’ACB présentent un risque accru d’endocardite et doivent recevoir des instructions comportementales appropriées.
- Le suivi cardiologique pour poser à temps l’indication d’un remplacement valvulaire et/ou aortique réduit le risque de complications graves (dissection aortique, dysfonctionnement cardiaque irréversible).
Littérature :
- Verma S, et al : Dilatation aortique chez les patients avec valve aortique bicuspide. N Engl J Med 2014 ; 370(20) : 1920-1929.
- Michelena HI, et al. : Incidence de l’endocardite infectieuse chez les patients porteurs de valves aortiques bicuspides dans la communauté. Mayo Clin Proc 2016 ; 91(1) : 122-123.
- Braverman AC, et al : La valve aortique bicuspide. Curr Probl Cardiol 2005 ; 30(9) : 470-522.
- Dijkema EJ, et al : Diagnostic, imagerie et prise en charge clinique de la coarctation aortique. Heart 2017 ; 103(15) : 1148-1155.
- Kwon MH, et al : Valvulopathie aortique bicuspide et aortopathie associée : une revue des études contemporaines pertinentes pour la prise de décision clinique. Curr Treat Options Cardiovasc Med 2017 ; 19(9) : 68.
- Debiec R, et al : Genetic Insights Into Bicuspid Aortic Valve Disease. Cardiol Rev 2017 ; 25(4) : 158-164.
- Evangelista A, et al : Insights From the International Registry of Acute Aortic Dissection : A 20-Year Experience of Collaborative Clinical Research. Circulation 2018 ; 137(17) : 1846-1860.
CARDIOVASC 2018 ; 17(3) : 20-24









