Nuove terapie sotto forma di anticorpi specifici e “piccole molecole” hanno inaugurato una nuova era. I biologici intervengono nel processo infiammatorio della dermatite atopica influenzando in modo specifico singole citochine. Gli inibitori di JAK, invece, sono farmaci a piccole molecole che mirano all’inibizione della via di segnalazione JAK/STAT. Nel senso della medicina personalizzata, si tratta sempre più di scegliere il farmaco più adatto all’individuo.
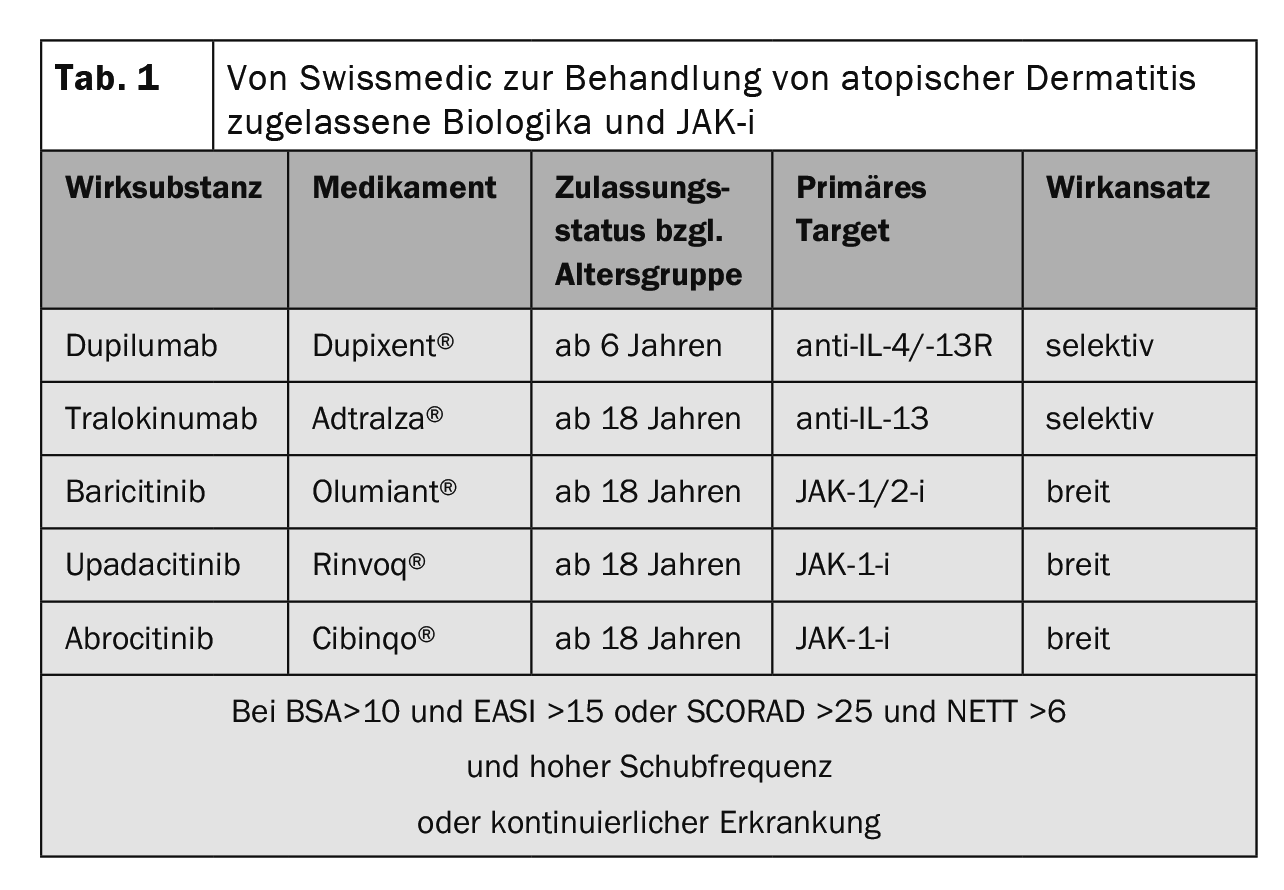
La complessa fisiopatologia della dermatite atopica (AD) coinvolge la predisposizione genetica, i fattori ambientali e la disregolazione dell’immunità innata e adattativa. Le risposte immunitarie di tipo 2 non solo rappresentano il correlato cellulare dell’infiammazione cutanea nell’AD, ma influenzano anche in modo significativo la disfunzione della barriera e la disbiosi microbica [1,2]. Il punteggio di gravità della dermatite atopica (SCORAD) può essere utilizzato per definire la gravità della DA. Al di sotto dei 25 punti l’eczema atopico viene classificato come lieve, tra i 25 e i 60 punti come moderato e al di sopra dei 60 come grave [3,4]. In circa un quinto dei casi di AD, i decorsi sono da moderati a gravi. Gli obiettivi terapeutici generali comprendono la riduzione del prurito, il miglioramento della risposta infiammatoria e il ripristino di una barriera cutanea intatta [5]. Le linee guida internazionali raccomandano che il trattamento dell’AD sia adattato individualmente alle diverse fasi, alla gravità e alla cronicità, secondo lo schema della terapia a tappe [6]. Negli ultimi anni, ci sono stati progressi significativi, soprattutto nell’ambito delle opzioni di trattamento sistemico [7]. Oltre agli anticorpi monoclonali tralokinumab e dupilumab somministrati come iniezioni sottocutanee, gli inibitori della Janus chinasi (JAK) baricitinib, upadacitinib e abrocitinib sono disponibili in forma di dosaggio orale (Tabella 1) .
I biologici attualmente approvati – IL-4 e IL-13 come citochine chiave
Le citochine Th2 interleuchina (IL)-4 e IL-13 sono significativamente coinvolte nelle reazioni allergiche IgE-mediate e svolgono un ruolo fisiopatologico chiave nelle risposte infiammatorie e nella disfunzione della barriera nell’AD [10,14,25]. Un ciclo di feedback positivo determina una riduzione della differenziazione dei cheratinociti e un aumento dell’iperplasia epidermica.
Dupilumab blocca in modo specifico le vie di segnalazione IL-4/IL-13 ed è il primo anticorpo monoclonale approvato nell’area di indicazione della dermatite atopica. In Svizzera, dupilumab (Dupixent®) è stato approvato per il trattamento della dermatite atopica da moderata a grave negli adulti dal 2019, seguito poi da un’estensione dell’indicazione per gli adolescenti e, da giugno 2022, dupilumab può essere utilizzato anche per i bambini a partire dai 6 anni [24].
Negli adulti, la dose iniziale raccomandata è di 600 mg di dupilumab (due iniezioni da 300 mg) e la dose di mantenimento è di 300 mg ogni due settimane. Per i bambini e gli adolescenti dai 6 ai 17 anni, il dosaggio consigliato dipende dal peso corporeo.
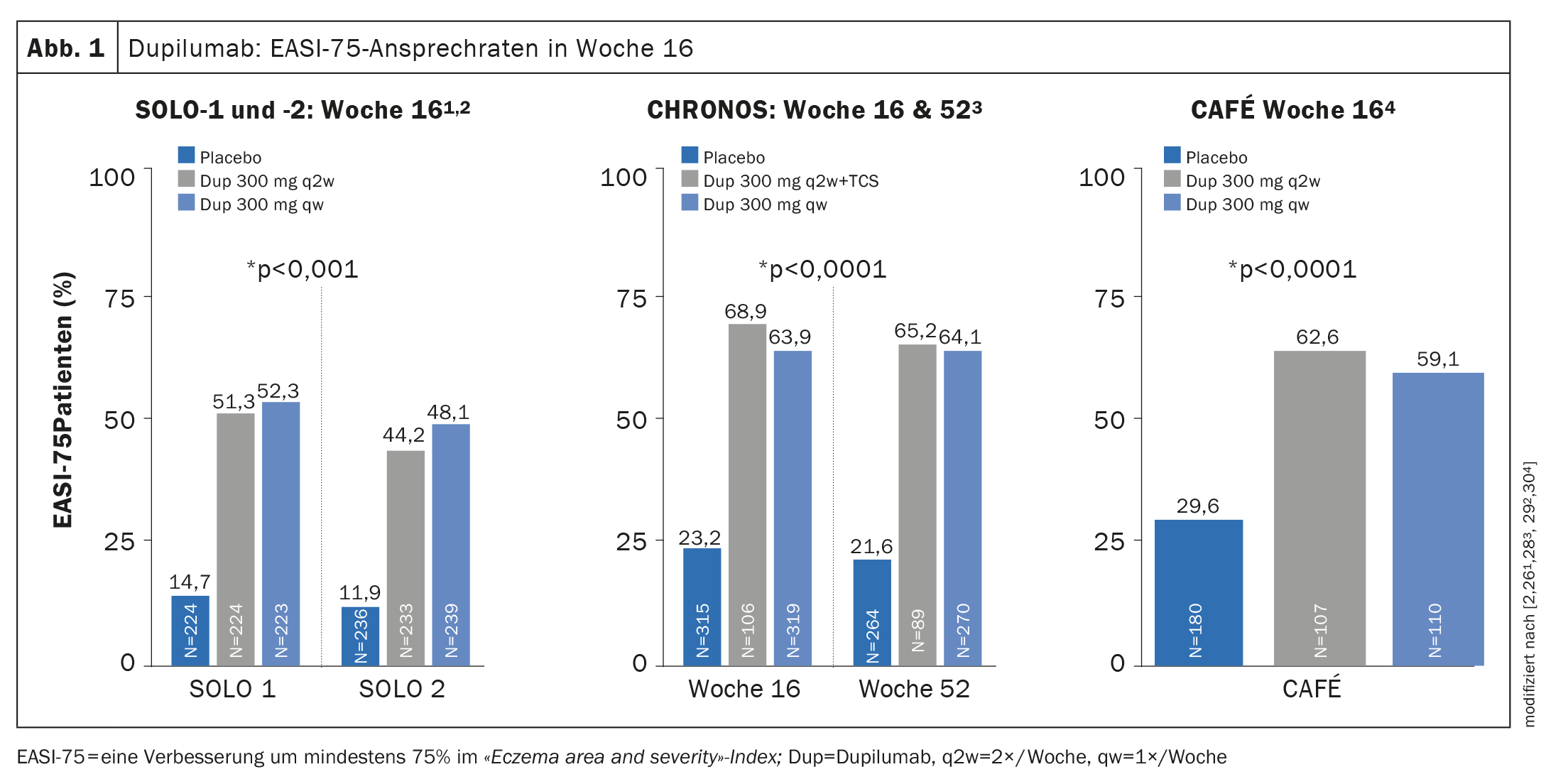
Prove di efficacia negli studi di fase III: sia in SOLO-1 e -2 (monoterapia) che in CHRONOS (terapia combinata con TCS concomitante), oltre il 35% dei pazienti ha raggiunto un punteggio Investigator’s Global Assessment (IGA) di 0 o 1 alla settimana 16, corrispondente a una pelle senza aspetto o quasi senza aspetto [26,28]. Il tasso di risposta EASI-75 è stato raggiunto sia in SOLO-1 e -2 che in CAFÉ alla settimana 16 (Fig. 1) e in CHRONOS anche alla settimana 52 [26,28–30]. Nello studio OLE, il tasso di risposta IGA 0/1 ha raggiunto un plateau di circa il 60% alla settimana 48, che è stato mantenuto fino all’endpoint dello studio alla settimana 76 [31,43].
Inoltre, dupilumab ha mostrato un rapido sollievo dal prurito in un confronto con placebo, con un miglioramento dei valori medi dei minimi quadrati di circa il 20% entro due settimane dall’inizio del trattamento [26,28–31].
L’approvazione di dupilumab per gli adolescenti si basa sui dati clinici del programma LIBERTY AD. L’efficacia e la sicurezza della monoterapia con dupilumab alla dose di 200 mg o 300 mg ogni due settimane sono state valutate in uno studio randomizzato, in doppio cieco e controllato su 251 pazienti di età compresa tra 12 e 17 anni con AD da moderata a grave [32].
Con dupilumab, il 42% ha ottenuto una risposta EASI-75 e il 24% ha ottenuto una IGA 0/1. Nel gruppo placebo, i valori corrispondenti erano 8% e 2%. Per la popolazione pediatrica di 6-11 anni, l’efficacia e la sicurezza di dupilumab in combinazione con i corticosteroidi topici (TCS) sono state analizzate negli studi AD-1652 e AD-1434 [33].
Profilo di sicurezza: gli effetti collaterali più comuni di dupilumab includono reazioni al sito di iniezione e congiuntivite, oltre a dolori articolari, herpes orale ed eosinofilia [33]. La congiuntivite può essere trattata con successo nella maggior parte dei casi con farmaci topici (acido ialuronico o fluorometolone (0,1%) collirio o tacrolimus (0,03) pomata oculare) [34]. I segnali di sicurezza di dupilumab nei bambini e negli adolescenti sono risultati ampiamente comparabili a quelli degli adulti.
Controindicazioni: Ad eccezione di rarissime reazioni di ipersensibilità sistemica generale, non sono note controindicazioni [24].
Monitoraggio della terapia e precauzioni: Si raccomanda di aggiornare lo stato di vaccinazione dei pazienti in base alle attuali raccomandazioni di vaccinazione prima di iniziare il trattamento con Dupilumab [24]. I vaccini vivi e i vaccini vivi attenuati non devono essere utilizzati in concomitanza con dupilumab, poiché la sicurezza e l’efficacia clinica non sono state stabilite.
Il tralokinumab è un anticorpo monoclonale umano che neutralizza specificamente l’IL-13. In Svizzera, il tralokinumab (Adtralza®) è stato approvato dal giugno 2022 per i pazienti adulti con MA da moderata a grave quando la terapia con farmaci topici non è efficace [24].
La dose iniziale per gli adulti è di 600 mg (4 iniezioni da 150 mg ciascuna), seguita da una dose di mantenimento di 300 mg (2 iniezioni da 150 mg ciascuna), con un intervallo di dosaggio di 2 settimane ciascuna. Dalla 16esima settimana, l’intervallo può essere esteso a 4 settimane.
A differenza di dupilumab, tralokinumab non si lega al recettore, ma alla parte solubile dell’IL-13, impedendo così l’interazione con il recettore IL-13Rα1 [35,36]. L’IL-13 è una citochina chiave nella patogenesi della dermatite atopica [35]. La sovraespressione dell’IL-13 è stata riscontrata sia nella pelle atopica lesionale che in quella non lesionale e i livelli di IL-13 sono stati trovati in correlazione con la gravità dell’AD [37].
Prove di efficacia negli studi di fase III: in ECZTRA-1 e -2, il 15,8% e il 22,2%, rispettivamente, dei pazienti trattati ogni due settimane (q2w) con tralokinumab 300 mg hanno raggiunto l’IGA 0/1 (senza lesioni o quasi) alla settimana 16, rispetto a solo il 7,1% e il 10,9%, rispettivamente, nel gruppo di confronto con placebo [39,40]. L’EASI-75 è stato raggiunto dal 25% dei partecipanti allo studio trattati con tralokinumab 300 mg q2w alla settimana 16 in ECZTRA-1 e la percentuale corrispondente era del 33,2% in ECZTRA-2 (Fig. 2) [39,40].
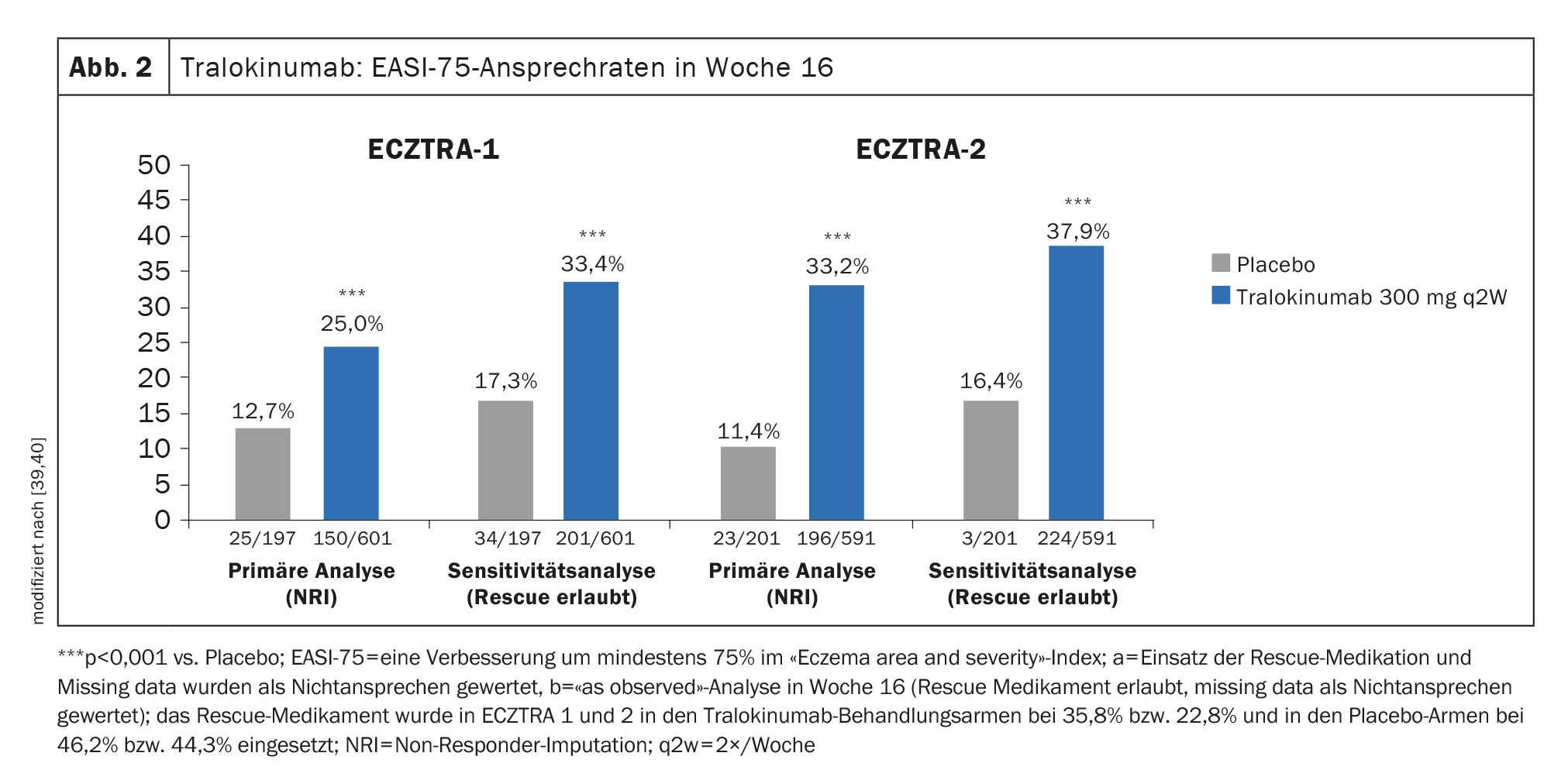
In ECZTRA-3, tralokinumab 300 mg q2w più TCS ha raggiunto l’EASI-75 alla settimana 16, con il 56,0% dei partecipanti allo studio che ha raggiunto l’EASI-75 rispetto al placebo, dove questo tasso era del 35,7%. Per quanto riguarda l’IGA 0/1, il braccio tralokinumab si è dimostrato significativamente superiore anche nel confronto con il placebo (38,9% vs. 26,2%) [37].
Profilo di sicurezza: gli effetti collaterali più comuni sono l’infezione del tratto respiratorio superiore/freddo e le reazioni al sito di iniezione. La congiuntivite si è verificata con una frequenza leggermente inferiore rispetto a quella di dupilumab [41].
Controindicazioni: Ad eccezione di rarissime reazioni di ipersensibilità sistemica generale, non sono note controindicazioni [24].
Monitoraggio della terapia e precauzioni: I vaccini vivi e i vaccini vivi attenuati non devono essere somministrati in concomitanza con tralokinumab, poiché la sicurezza e l’efficacia clinica non sono state stabilite. Gli intervalli di tempo tra una vaccinazione con vaccini vivi e il trattamento con tralokinumab devono essere ricavati dalle attuali raccomandazioni di vaccinazione.
Anche gli inibitori della JAK sono in aumento.
Gli inibitori della Janus chinasi (JAK) sopprimono la risposta intracellulare alle citochine (meccanismo JAK-STAT). Il meccanismo d’azione degli inibitori JAK consiste nell’utilizzare “piccole molecole” per bloccare le JAK su cui si basano i recettori delle citochine. Con baricitinib, upadacitinib e abrocitinib, tre JAK-i somministrabili per via orale sono attualmente approvati in Svizzera per il trattamento sistemico dell’AD da moderata a grave [24]. Le citochine patogeneticamente centrali che avviano la risposta citochinica attraverso la via JAK-STAT sono IL-4, IL-13 e IL-31 [44].
L’aspetto particolarmente interessante di JAK-i come opzione terapeutica è la sua rapida insorgenza d’azione. Il profilo degli effetti collaterali di JAK-i è dipendente dalla sostanza e dalla dose, con infezioni che si verificano più frequentemente rispetto ai biologici e alcuni parametri di laboratorio che vengono alterati durante la terapia, per cui è necessario un monitoraggio intensificato dei pazienti.
Baricitinib è un inibitore orale selettivo di JAK1 e JAK2. Attraverso JAK1, vengono trasmessi i segnali delle citochine chiave adattative e innate IL-4, IL-13, IL-22, IL-31, TSLP, IFN-γ e IL-6 e JAK2 è necessario per la funzione del recettore IL-5, richiesto per il reclutamento dei granulociti eosinofili [1].
Il baricitinib (Olumiant®) è stato approvato in Svizzera dal febbraio 2021 per il trattamento di pazienti adulti con dermatite atopica da moderata a grave [24,45]. Il dosaggio standard è di 4 mg (1×/d p.o.). La dose di 2 mg (1×/d p.o.) è presa in considerazione nei pazienti di 75 anni e oltre o nei pazienti con infezioni croniche/ricorrenti o con una clearance della creatinina di 30-60 ml/min. Se non è evidente alcun beneficio terapeutico di baricitinib dopo 8 settimane, si deve prendere in considerazione l’interruzione della terapia.
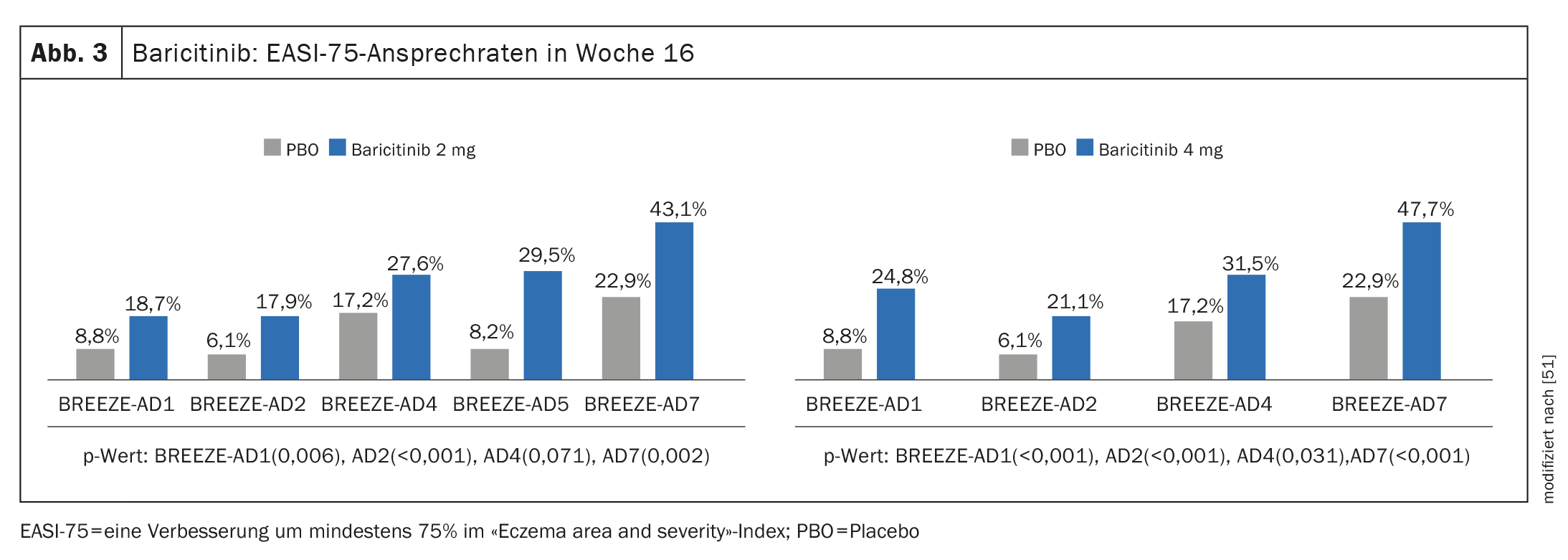
Prova di efficacia negli studi di fase III: oltre a BREEZE-AD1 e BREEZE-AD2, in cui la monoterapia con baricitinib ha determinato una riduzione significativa dei sintomi, il programma di studi cardine di fase III comprendeva anche BREEZE-AD7, per studiare l’efficacia e la sicurezza di baricitinib in combinazione con corticosteroidi topici (TCS) [45–47]. 329 pazienti adulti con MA da moderato a grave sono stati randomizzati in BREEZE-AD7 in un rapporto 1:1:1 a baricitinib 4 mg più TCS o baricitinib 2 mg più TCS o placebo. Alla settimana 16, circa il 47,7% dei pazienti che hanno ricevuto baricitinib 4 mg in combinazione con TCS ha mostrato una risposta EASI-75, rispetto al 22,9% del gruppo placebo (p<0,001) (Fig. 3). Nel braccio con baricitinib 2 mg, i valori corrispondenti erano rispettivamente 43,1% e 22,9%. (Fig. 3).
Un miglioramento del prurito di ≥4 punti sulla scala di valutazione numerica (NRS) entro le prime 16 settimane è stato ottenuto nel 44% dei pazienti trattati con baricitinib (4 mg più TCS), rispetto al 20% con placebo più TCS (p<0,01) [48]. Sono disponibili dati di studio completi sulla tollerabilità di baricitinib nell’AD, provenienti da un totale di 8 studi randomizzati che hanno coinvolto 2531 pazienti e 2247 anni-paziente [49].
Profilo di sicurezza: le reazioni avverse più comunemente riportate durante la terapia con baricitinib sono l’aumento del colesterolo LDL, le infezioni del tratto respiratorio superiore, la cefalea, l’herpes simplex e le infezioni del tratto urinario [50]. L’incidenza di infezioni gravi con baricitinib era simile a quella del placebo.
Controindicazioni: Baricitinib è controindicato durante la gravidanza. Le donne in età fertile devono utilizzare un metodo contraccettivo affidabile durante l’uso di baricitinib e per almeno un’altra settimana dopo la fine del trattamento Nei pazienti con grave compromissione epatica o con una clearance della creatinina <30 ml/min, l’uso di baricitinib non è raccomandato [50].
Monitoraggio della terapia e precauzioni: Prima di iniziare il trattamento, i pazienti devono essere valutati per la presenza di disfunzioni epatiche e renali gravi e di fattori di rischio per trombosi venosa profonda o embolia polmonare. Anche i livelli lipidici (colesterolo totale, HDL, LDL, trigliceridi) devono essere valutati e monitorati 12 settimane dopo o secondo la linea guida sull’iperlipidemia. Inoltre, i pazienti devono essere sottoposti al test per la tubercolosi (TB) prima di iniziare la terapia con baricitinib. Baricitinib non deve essere utilizzato nella TBC attiva, e nella TBC latente non trattata in precedenza, si deve prendere in considerazione la terapia anti-TB prima di iniziare il trattamento con baricitinib. Se un paziente sviluppa un’infezione da herpes zoster, il trattamento con baricitinib deve essere temporaneamente interrotto fino alla guarigione dell’infezione. Lo screening per l’epatite virale deve essere effettuato prima di iniziare la terapia con baricitinib. I pazienti con infezione attiva accertata da epatite B o epatite C sono stati esclusi dagli studi clinici. Si raccomanda di aggiornare tutte le immunizzazioni in conformità con le attuali raccomandazioni di vaccinazione prima della terapia con baricitinib. L’uso di vaccini vivi attenuati non è raccomandato durante o immediatamente prima del trattamento con baricitinib.
Upadacitinib è un inibitore della Janus chinasi peroralmente biodisponibile, selettivo e reversibile che inibisce principalmente JAK1. In Svizzera, upadacitinib (Rinvoq®) è stato approvato per il trattamento dell’AD da moderato a grave negli adulti dal novembre 2021 [52]. La dose orale raccomandata è di 15 mg di upadacitinib una volta al giorno.
Prove di efficacia negli studi di fase III: l’efficacia di upadacitinib nell’AD è stata analizzata negli studi di fase III MEASURE Up-1, MEASURE Up-2 e AD Up, con un totale di 2584 pazienti inclusi nel programma di studio. In tutti e tre gli studi, i partecipanti hanno ricevuto 15 mg o 30 mg di upadacitinib o placebo una volta al giorno per 16 settimane e il miglioramento significativo dei punteggi SCORAD e EASI è stato dimostrato nel confronto con il placebo. (Fig. 4). Il miglioramento dell’aspetto della pelle e del prurito è stato più rapido. [52].
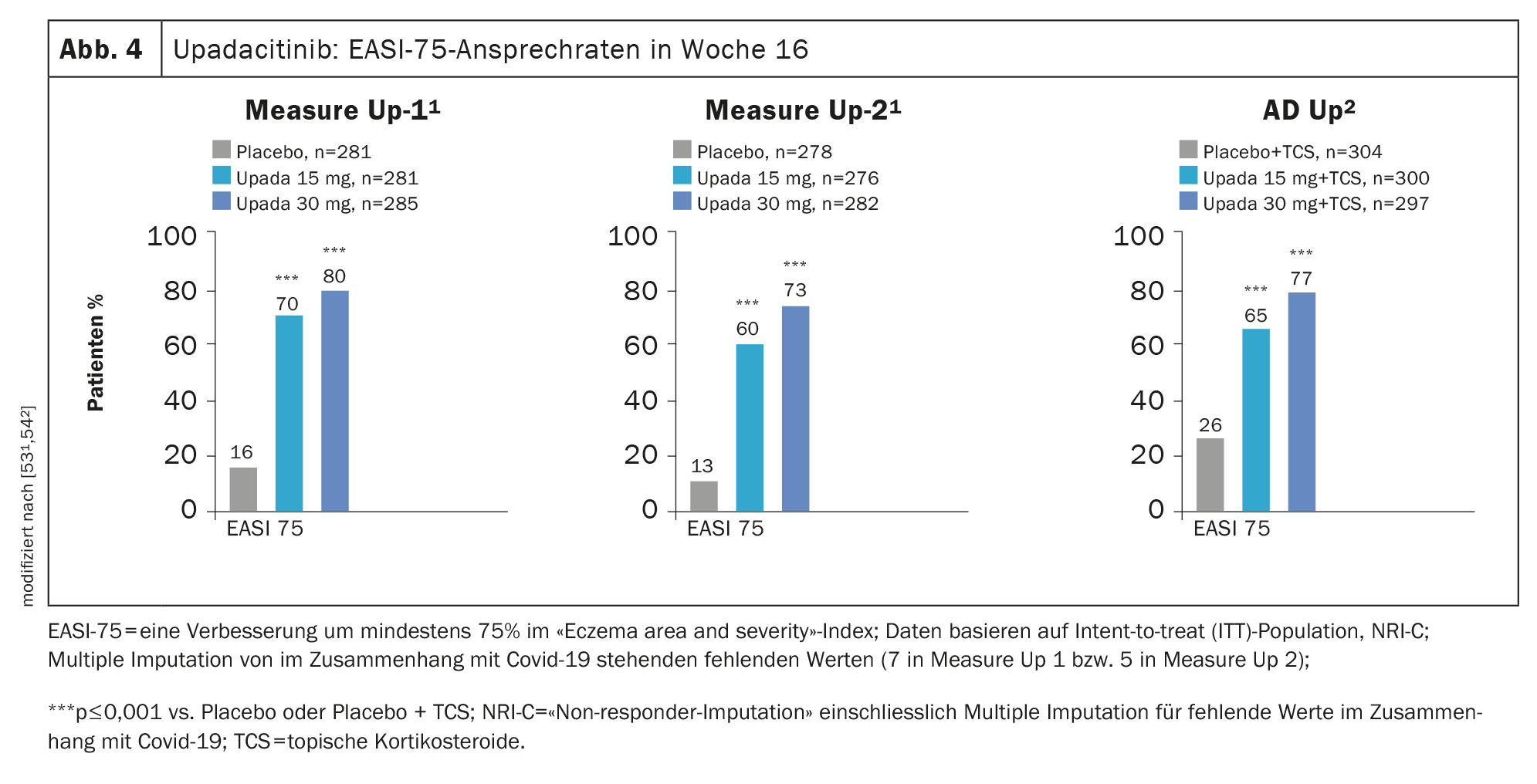
In MEASURE Up-1 e -2, upadacitinib è stato studiato come monoterapia nell’AD da moderato a grave in una popolazione di 1683 pazienti. Dopo 16 settimane, oltre il 40% ha ottenuto una risposta EASI 90 e un significativo sollievo dal prurito con upadacitinib 15 mg (1×d), mentre nei gruppi placebo entrambe le cose sono state riscontrate solo in circa un paziente su dieci [53,54]. In un’analisi integrata di entrambi gli studi Measure Up-1 e Measure Up-2, upadacitinib ha mostrato una risposta EASI-90 del 62% e un miglioramento significativo del prurito del 65% alla settimana 52. [55]. Una risposta EASI-75 è stata raggiunta da circa l’80% [55]. Inoltre, il trattamento con upadacitinib è stato associato a una risposta rapida: un miglioramento del prurito è stato osservato dopo soli 2 giorni .
Profilo di sicurezza: il profilo degli effetti collaterali di upadacitinib è simile a quello di altri inibitori JAK. Le più comuni sono le infezioni del tratto respiratorio superiore e l’acne, meno comuni sono l’herpes simplex, le emicranie e l’aumento della creatina fosfochinasi nel sangue [52].
Controindicazioni: Upadacitinb è controindicato durante la gravidanza; le donne in età fertile devono utilizzare metodi contraccettivi affidabili sia durante il trattamento che fino a quattro settimane dopo l’ultima dose di upadacitinib. Si raccomanda di non utilizzare upadacitinib nei pazienti con una conta assoluta dei linfociti inferiore a 500 cellule/mm3, una conta assoluta dei neutrofili inferiore a 1000 cellule/mm3 o un livello di emoglobina inferiore a 8 g/dl [24].
Non è necessario alcun aggiustamento della dose nei pazienti con compromissione epatica lieve o moderata. Nei casi di grave compromissione epatica (Child-Pugh C), l’uso di upadacitinib non è raccomandato. L’insufficienza renale non ha un’influenza clinicamente rilevante sull’esposizione di upadacitinib [52].
Monitoraggio della terapia e precauzioni: Lo screening della tubercolosi (TB) deve essere effettuato prima dell’inizio della terapia con upadacitinib. Upadacitinib non deve essere utilizzato nei pazienti con TB attiva. Nei pazienti con TB latente non trattata, la terapia anti-TB deve essere presa in considerazione prima di iniziare il trattamento con upadacitinib. Se un paziente sviluppa l’herpes zoster, si deve prendere in considerazione la possibilità di interrompere il trattamento con upadacitinib fino alla guarigione dell’infezione. Prima di iniziare e durante la terapia con upadacitinib, è necessario effettuare lo screening per l’epatite virale e il monitoraggio della possibile riattivazione. I pazienti che sono risultati positivi agli anticorpi dell’epatite C e all’RNA del virus dell’epatite C sono stati esclusi dagli studi clinici.
Prima di iniziare la terapia con upadacitinib, si raccomanda di controllare lo stato di vaccinazione dei pazienti secondo le attuali linee guida di vaccinazione e di recuperare tutte le vaccinazioni richieste. L’uso di vaccini vivi attenuati durante o immediatamente prima del trattamento con upadacitinib non è raccomandato [52].
Abrocitinib è un inibitore selettivo di JAK-1 per via orale. In Svizzera, abrocitinib (Cibinqo®) è stato approvato per il trattamento della dermatite atopica negli adulti dall’aprile 2022 [24].
Prove di efficacia negli studi di fase III: L’efficacia e la sicurezza di abrocitinib come monoterapia e in combinazione con terapie di fondo attive applicate per via topica per un periodo di 12-16 settimane sono state analizzate in 1616 pazienti negli studi di fase III controllati con placebo MONO-1, MONO-2 e COMPARE. Nello studio controllato e randomizzato REGIMEN, che è rilevante anche per l’approvazione, abrocitinib è stato studiato per un periodo di 52 settimane in un totale di 1233 partecipanti allo studio [56].
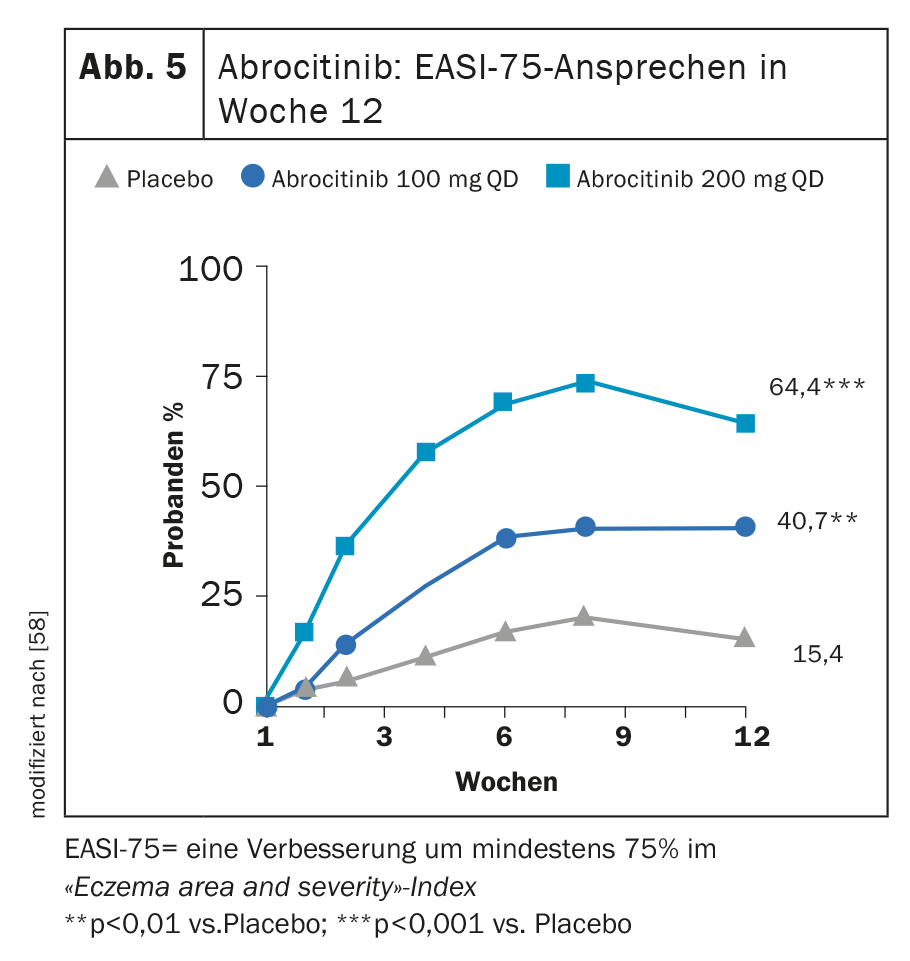
In MONO-1 e MONO-2, il trattamento con abrocitinib 100 mg (1× d) ha raggiunto entrambi gli endpoint primari IGA 0 o 1 e/o EASI-75 in una percentuale significativamente maggiore alla settimana 12 (Fig. 5) [24]. In combinazione con la TCS, l’inibitore della JAK si è dimostrato significativamente superiore al placebo nello studio COMPARE alla settimana 16 per quanto riguarda questi due endpoint. Una riduzione del prurito di ≥4 punti sul NRS del prurito si è manifestata con abrocitinib rispetto al placebo già alla settimana 2 in una percentuale significativamente maggiore di partecipanti allo studio. [57]. Alla dose elevata di 200 mg, il 43,8% dei pazienti ha mostrato un miglioramento dell’IGA e il 64,6% un EASI-75 [58] dopo 12 settimane in uno studio di fase IIb.
Profilo di sicurezza: gli effetti collaterali più comunemente riportati sono nausea, mal di testa, acne, herpes simplex, aumento della creatina fosfochinasi nel sangue, vomito, vertigini e dolore addominale superiore. Gli effetti collaterali gravi più comuni (0,3%) sono le infezioni [56].
Controindicazioni: Abrocitinib è controindicato durante la gravidanza. Le donne in età fertile devono essere avvisate di utilizzare una contraccezione efficace durante il trattamento e per un mese dopo la fine della somministrazione perorale di upadacitinib. Abrocitinib è controindicato in caso di grave compromissione epatica (classe C di Child-Pugh). Non è necessario alcun aggiustamento della dose nei casi di lieve insufficienza renale. In caso di insufficienza renale moderatamente grave (eGFR da 30 ml/min a <60 ml/min o eGFR <30 ml/min), la dose deve essere ridotta in conformità alle informazioni sul prodotto [56].
Monitoraggio della terapia e precauzioni: I pazienti devono essere sottoposti a un test per la tubercolosi (TB) prima di iniziare il trattamento con abrocitinib. Abrocitinib non deve essere somministrato a pazienti con TB attiva. Nei pazienti con TB latente non trattata, la terapia preventiva per la TB latente deve essere iniziata prima di iniziare il trattamento.
Se un paziente sviluppa un’infezione da herpes zoster, si deve prendere in considerazione la possibilità di interrompere temporaneamente il trattamento con abrocitinib fino alla risoluzione dell’infezione. Prima di iniziare e durante la terapia, è necessario eseguire uno screening per l’epatite virale, secondo le linee guida cliniche. I pazienti con infezione attiva accertata da epatite B o epatite C sono stati esclusi dagli studi clinici. Inoltre, si raccomanda di aggiornare tutte le immunizzazioni in conformità alle attuali raccomandazioni di vaccinazione prima di iniziare il trattamento con abrocitinib. L’uso di vaccini vivi attenuati deve essere evitato durante o immediatamente prima del trattamento con abrocitinib [56].
Altri composti promettenti sono in cantiere
Altri principi attivi attualmente in fase di ricerca per l’uso nell’AD includono [2]:
- Lebrikizumab: anticorpo monoclonale che si lega specificamente all’IL-13 liberamente circolante. Oltre ai due studi di monoterapia di fase III ADvocate 1 e 2, la combinazione di lebrikizumab con TCS viene studiata in ADhere [62].
- Nemolizumab: anticorpo monoclonale che blocca la subunità α del recettore IL-31. I dati degli studi condotti finora mostrano un forte sollievo dal prurito in particolare [63]. In uno studio randomizzato di 16 settimane con randomizzazione 2:1, alla settimana 16 il gruppo nemolizumab (n=143) ha mostrato una variazione percentuale media del punteggio VAS di -42,8% rispetto a -21,4% del placebo (n=72) [64].
- Tezepelumab: anticorpo contro la TSLP (linfoproteina stromale timica). La TSLP è una citochina derivata dalle cellule epiteliali che è particolarmente associata al prurito nell’AD [65]. Nello studio ALLEVIAD di fase IIa, il miglioramento percentuale medio del picco di prurito NRS di tezelumab più TCS rispetto al placebo alla settimana 12 è stato di 33,54 (SD 6,02) vs 25,41 (SD 6,06); p=0,258) [68].
- anticorpo anti-Ox-40: Ox-40 è un recettore costimolatorio sulle cellule T attivate [65]. In uno studio di fase IIa, randomizzato, in doppio cieco e controllato con placebo, la somministrazione dell’anticorpo anti Ox-40 a intervalli di 4 settimane ha portato a miglioramenti clinici significativi entro il giorno 71 [70].
- Brepocitinib: inibitore topico di TYK2/JAK1
- Modulatori dei recettori degli idrocarburi arilici: sistema recettoriale citoplasmatico che influenza il meccanismo infiammatorio e le molecole di barriera.
Messaggi da portare a casa
- Le nuove terapie sotto forma di anticorpi specifici e le cosiddette piccole molecole hanno inaugurato una nuova era. L’interleuchina (IL-)4 e l’IL-13 sono le due citochine pro-infiammatorie più importanti nell’AD. Inoltre, ci sono altri mediatori infiammatori che svolgono un ruolo.
- I biologici intervengono nel processo infiammatorio nell’AD influenzando in modo specifico singole citochine. Gli inibitori di JAK, invece, sono farmaci a piccole molecole che mirano all’inibizione della via di segnalazione JAK/STAT.
- Quando una citochina si lega al dominio extracellulare del suo recettore, le Janus chinasi attivano i fattori di trascrizione intracellulari che controllano la lettura di un gran numero di geni nel nucleo cellulare [1].
- Le differenze più significative tra i moderni terapici sistemici altamente efficaci riguardano la forma di somministrazione, la frequenza d’uso, il profilo degli effetti collaterali e il monitoraggio della terapia. Nel senso della medicina personalizzata, si tratta sempre più di scegliere il farmaco più adatto alle caratteristiche della malattia e ad altre caratteristiche legate alla persona e al contesto.
Letteratura:
- Lauffer F, Biedermann T: Dtsch Arztebl 2021; 118(24): [24]; DOI: 10.3238/PersDerma.2021.06.18.04
- “Dermatite atopica, cosa c’è di nuovo?”, Univ.-Prof. Dr. Paul-Gunther Sator, DermAlpin Salzburg, 30-31.10.2021
- Deutsche Haut- und Allergiehilfe e.V, www.dha-neurodermitis.de/therapie/grundlagen-der-therapie.html,(ultimo accesso, 06.01.2023)
- Punteggio di gravità della dermatite atopica: l’indice SCORAD. Dermatologia 1993; 186(1): 23-31.
- Nygaard U, Deleuran M, Vestergaard C: Dermatologia 2017; 233: 333-343.
- AWMF: Linea guida Neurodermite, numero di registro 013-027, classificazione S2k Stato: 31.03.2015.
- Studio di letteratura DFP: Dermatite atopica, www.oeadf.at/files/E-Learning/ClinicumDerma_12_032017.pdf,(ultimo accesso 06.01.2023)
- Leung DYM, Guttman-Yassky E: J Allergy Clin Immunol 2014; 134: 769-779.
- Kim BE, Leung DY, Boguniewicz M, Howell MDl: Clin Immunol 2008; 126: 332-337.
- Akdis CA, et al: Allergy 2020; 75(7): 1582-1605.
- Hoffjan S, Stemmer S: Arch Dermatol Res 2015; 307: 659-670.
- Darsow U, et al: J Eur Acad Dermatol Venereol 2010; 24: 317-328.
- Williams MR, Gallo RL: Curr Allergy Asthma Rep 2015; 15: 65.
- Nguyen JK, et al: Arch Dermatol Res 2020; 312(2): 81-92.
- Weidinger S, et al: Nat Rev Dis Primers 2018; 4(1): 2.
- Clark JD, et al: J Med Chem 2014; 57: 5023-5038.
- Howell MD, et al: Front Immunol 2019; 10: 2342.
- Chiesa Fuxench ZC, Ong P: Poster presentato all’AAD 2018. Poster 6236.
- Drucker AM, et al: J Invest Dermatol 2017; 137(1): 26-30.
- Ronnstad ATM, et al: JAAD 2018; 79: 448-456.
- Reich K, et al: Società Internazionale di Dermatite Atopica 2021.
- Brunner PM, et al: J Invest Dermatol 2017; 137: 18-25.
- Silverberg JI: Ann Allergy Asthma Immunol 2019; 123(2): 144-151.
- Informazioni sul soggetto, www.swissmedicinfo.ch, (ultimo accesso 06.01.2022)
- 25 Silverberg J: Clin Dermatol 2017; 35: 360-366.
- Simpson EL, et al: N Engl J Med 2016; 375: 2335-2348.
- Paller AS et al: J Am Acad Dermatol. 2016; 75(3): 494-503.
- Blauvelt A, et al: The Lancet 2017; 389(10086): 2287-2303.
- Simpson EL, et al. Abstract dell’ultima ora, abstract EADV 2016 D3T01.1C.
- de Bruin-Weller, M. et al: Br J Dermatol 2018; 178(5): 1083-1101.
- Deleuran M et al: J Am Acad Dermatol 2020; 82(2): 377-388.
- Simpson EL, et al: JAMA Dermatol 2020; 156(1): 44-56.
- Agenzia Europea dei Medicinali: Dupilumab, Informazioni sul prodotto, https://ec.europa.eu/healthhttps://ec.europa.eu/health, ultimo accesso 06.01.2023)
- Wollenberg A, et al: J Allergy Clin Immunol Pract. 2018 Sep-Oct; 6(5): 1778-1780.e1.
- Schmid-Grendelmeier P: Dermatologie Praxis 2021; 31(4): 10-14.
- Wollenberg A, et al: JDDG 2021; 19(10): 1435-1442.
- Silverberg JI, et al: Br J Dermatol 2021; 184: 450-463.
- Petronelli M: Tralokinumab soddisfa gli endpoint di tutti gli studi di fase 3, Dermatology Times, 16 dicembre 2019, www.dermatologytimes.com, (ultimo accesso 06 gennaio 2023)
- Simpson E, et al.: Presentazione all’AAD VMX, 12-14 giugno e on-demand, 2020, USA.
- Wollenberg A, et al: Br J Dermatol 2021; 184(3): 437-449.
- Agenzia Europea dei Medicinali, Tralokinumab, Informazioni sul prodotto,https://ec.europa.eu/healthhttps://ec.europa.eu/health, ultimo accesso 06.01.2023)
- “Giornata mondiale della neurodermite il 14 settembre Neurodermite: le terapie sistemiche migliorano il trattamento”, Berufsverband der Deutschen Dermatologen e.V. (BVVD), Berlino 03.09.2021.
- Futamura MJ: Am Acad Dermatol 2016; 74: 288-294.
- Klein B, Treudler R, Simon JC: JDDG 2022; 20(1): 19-25.
- Simpson EL, et al. Presentato al:24° Congresso Mondiale di Dermatologia; 10-15 giugno 2019; Milano, Italia.
- Melo A, Carrascosa JM, Torres T: J Dermatolog Treat 2021 Aug 23: 1-10. Pubblicato prima della stampa.
- Wollenberg A, et al: JEADV 2021; 35(7): 1543-1552.
- Reich K, et al: JAMA Dermatol 2020; 156(12): 1333-1343.
- Silverberg JI, et al: JAMA Dermatol 2021, 157: 691-699.
- Agenzia Europea dei Medicinali: Baricitinib, Informazioni sul prodotto, https://ec.europa.eu/health, ultimo accesso 06.01.2023)
- 51. radi G, et al: Healthcare (Basel) 2021 Nov 18; 9(11):1575. healthcare-09-01575-v2.pdf.
- Agenzia Europea dei Medicinali: Upadacitinib, Informazioni sul prodotto, https://ec.europa.eu/health, ultimo accesso 06.01.2023)
- Guttman-Yassky E, et al: Lancet 2021; 397(10290): 2151-2168.
- Reich K, et al: Lancet 2021; 397(10290): 2169-2181.
- Simpson EL, et al: Presentazione al 2021 Dermatology Education Foundation (DEF) Essential Resource Meeting (DERM2021), 5-8 agosto 2021, Las Vegas NV.
- Agenzia Europea dei Medicinali: Abrocitinib, Informazioni sul prodotto, https://ec.europa.eu/health/, (ultimo accesso 06.01.2023)
- “Dermatite atopica: la via di segnalazione JAK/STAT come bersaglio terapeutico ” https://medizinonline.com/der-jakstat-signalweg-als-therapeutische-zielstruktur,(ultimo accesso 16.02.2023).
- Gooderham MJ, et al: JAMA Dermatol 2019; 155(12): 1371-1379.
- Guttman-Yassky E, et al: JAMA Dermatol 2020; 156(4): 411-420.
- Moyle M, et al: Exp Dermatol 2019; 28(7): 756-768.
- 61. Loh TY, et al: J Asthma Allergy 2020; 13: 109-114.
- 62 “Eli Lilly presenta un BLA per il trattamento di Lebrikizumab AD”,
www.dermatologytimes.com/view/eli-lilly-submits-bla-for-lebrikizumab-ad-treatment,(ultimo accesso 06.01.2023) - Ruzicka T, et al: N Engl J Med 2017; 376: 826-835.
- Kabashima K, et al: N Engl J Med 2020; 383: 141-150.
- 65 Quint T, Bangert C: hautnah 2021; 20: 37-44.
- 66 Ziegler SF, et al: Adv Pharmacol 2013; 66: 129-155.
- Shikotra A, et al: J Allergy Clin Immunol 2012; 129: 104-111.e1-9.
- Simpson EL, et al: J Am Acad Dermatol 2019; 80: 1013-1021.
- Suga H, Sato S: Nuove terapie topiche e sistemiche nella dermatite atopica. Immunol Med 2019; 42(2): 84-93.
- Guttman-Yassky E, et al: J Allergy Clin Immunol 2019; 144(2): 482-493.e487.
- Freund Y, et al: FEBS Lett 2012; 586: 3410-3414.
- Dong C, et al: J Pharmacol Exp Ther 2016; 358: 413-422.
- Jarnagin K, et al: J Drugs Dermatol 2016; 15(4): 390-396.
- Zane LT, et al: Pediatr Dermatol 2016: 33(4): 380-387.
DERMATOLOGIE PRAXIS 2023; 30(1): 5–11