Le malattie da accumulo lisosomiale sono un gruppo eterogeneo di malattie genetiche che hanno origine in una disfunzione dei processi metabolici lisosomiali. La malattia di Niemann-Pick è oggi chiamata anche ASMD (“deficit di sfingomielinasi acida”). I tipi A e B sono classificati come sfingolipidosi, mentre il tipo C appartiene alle malattie da accumulo di lipidi. Per i tipi A e B, la prima terapia enzimatica sostitutiva è stata approvata nell’UE lo scorso anno.
La malattia di Niemann-Pick è una malattia genetica da accumulo lisosomiale che prende il nome dal pediatra tedesco Albert Niemann (1880-1921) e dal patologo tedesco Ludwig Pick (1868-1944). Ludwig Pick riuscì a distinguere la malattia di Niemann-Pick dalla malattia di Gaucher come malattia metabolica indipendente [1]. La sostanza di stoccaggio sfingomielina è stata scoperta dal biochimico Klenk nel 1934. Una classificazione in diversi sottogruppi è stata avviata da Crocker nel 1961 [2–4]. Le forme più comuni di manifestazione sono di tipo A-C (riquadro).
| I sottotipi più comuni La sindrome di Niemann-Pick è una malattia ereditaria autosomica recessiva. I tipi A e B sono causati da una carenza nell’attività di un enzima lisosomiale codificato dal gene SMPD1. Il difetto genetico significa che la sfingomielina non può più essere scomposta e si accumula nelle cellule di vari organi. Il tipo C è un disturbo del metabolismo del colesterolo in cui sono rilevabili mutazioni nel gene NPC-1 (18q11) o nel gene NPC-2 (14q24.3). |
| a [11] |
Manifestazioni cliniche
Il tipo A è una grave malattia neurodegenerativa dell’infanzia che di solito porta alla morte entro i primi tre anni di vita. I sintomi principali sono l’epatosplenomegalia e il declino psicomotorio. Lo sviluppo dei bambini colpiti ristagna. Le abilità apprese negli ultimi anni di vita si perdono nel tempo. Spesso, prima del completamento dei primi sei mesi di vita, si possono osservare insufficienza di crescita, vomito, perdita dell’udito, tetraspasticità e convulsioni miocloniche.
Il tipo B è caratterizzato da un esordio più tardivo della malattia e da una manifestazione più lieve rispetto al tipo A [5]. La maggior parte dei pazienti raggiunge l’età adulta e non si osservano sintomi cerebrali. Al contrario, sono caratteristiche l’epatosplenomegalia con ipersplenismo progressivo e la disfunzione epatica stabile, così come il graduale deterioramento della funzione polmonare, accompagnato da osteopenia e da un profilo lipidico aterogeno [6]. Un profilo lipidico pro-aterogeno è visibile all’inizio del decorso della malattia e alcuni pazienti sviluppano una malattia coronarica.
Il tipo C è associato a un’alterazione del trasporto del colesterolo dai lisosomi, con conseguente aumento dell’immagazzinamento di colesterolo, glicosfingolipidi e gangliosidi nei lisosomi di varie cellule del corpo [7,8]. Rispetto al tipo A/B, la presentazione clinica è molto eterogenea. Si tratta di una malattia neuroviscerale cronica che ha una progressione più lenta rispetto al tipo A. Può essere suddivisa in una forma infantile precoce, una forma infantile tardiva, una forma giovanile e una forma adulta [7]. Clinicamente, le persone colpite presentano varie anomalie neurologiche e psichiatriche, talvolta anche sintomi viscerali come l’epatosplenomegalia [8]. Le manifestazioni neurologiche tipiche del tipo C sono disturbi cognitivi, crisi epilettiche, anomalie comportamentali, depressione e psicosi, paralisi dello sguardo verticale, disturbi del linguaggio e della deglutizione e distonia [9].
| Tipo A/B: nuova terapia enzimatica sostitutiva: olipudase alfa Per i pazienti pediatrici e adulti con malattia di Niemann-Pick o ASMD (“deficit di sfingomielinasi acida”) di tipo A/B o di tipo B senza coinvolgimento del sistema nervoso centrale, la terapia enzimatica sostitutiva olipudase alfa è stata approvata dall’EMA nel 2022. L’Olipudase alfa è stata progettata per sostituire l’ASM mancante o difettoso per consentire la degradazione della sfingomielina. La decisione di approvazione si basa sui dati degli studi clinici ASCEND e ASCEND-Peds, che hanno mostrato miglioramenti clinicamente rilevanti della funzione polmonare e riduzioni del volume della milza e del fegato con la terapia con olipudase alfa. L’incidenza degli eventi avversi nei pazienti che hanno ricevuto olipudase alfa è stata paragonabile a quella del gruppo placebo. L’Olipudase alfa viene infuso ogni quindici giorni nella fase di mantenimento. Nello studio ASCEND, 36 pazienti adulti con ASMD di tipo A/B o di tipo B sono stati randomizzati a olipudase alfa o a placebo. Dopo 52 settimane, il braccio di trattamento ha mostrato un miglioramento della funzione polmonare e una riduzione del volume della milza. Nello studio ASCEND-PEDS a braccio singolo, 20 pazienti pediatrici con ASMD di tipo A/B o di tipo B sono stati trattati con olipudase alfa per 64 settimane. Anche in questo caso, gli endpoint più importanti sono stati raggiunti alla 52esima settimana. |
| a [12] |
Diagnostica e terapia
Nelle colture di leucociti e fibroblasti, è possibile rilevare l’attività ridotta o assente della sfingomielinasi acida; la causa della malattia di Niemann-Pick di tipo A e B è [10]. Questi esami genetici enzimatici e molecolari possono essere effettuati già in fase prenatale, se è nota una predisposizione familiare [1]. Per stabilire la diagnosi della malattia di Niemann-Pick di tipo C , devono essere eseguiti esami complessi del metabolismo del colesterolo [10]. I difetti genetici sottostanti non sono attualmente trattabili (a partire dal 2022). La terapia enzimatica sostitutiva con olipudase alfa è disponibile nell’UE per la malattia di Niemann-Pick di tipo A e B (riquadro). Il tipo C viene trattato sintomaticamente con miglustat.
Letteratura:
- “Esame del fegato e della milza mediante elastografia in pazienti affetti dalla malattia di Niemann-Pick di tipo B”, Gözde Aksu, tesi inaugurale, 2020, https://openscience.ub.uni-mainz.de,(ultimo accesso 12.10.2023).
- Crocker AC, Mays VB: Sintesi della sfingomielina nella malattia di Niemann-Pick. Am J Clin Nutr 1961; 9: 63-67.
- Crocker AC: Il difetto cerebrale nella malattia di Tay-Sachs e nella malattia di Niemann-Pick. J Neurochem 1961; 7: 69-80.
- E. K. Sulla natura dei fosfatidi della milza nella malattia di Niemann-Picksen. Hoppe Seyler’s Journal of Physiological Chemistry. 1934.
- Tran C, et al: Coinvolgimento polmonare nei pazienti adulti con errori innati del metabolismo. Karger Compass Pneumol 2018; 6: 6-17.
- Orphanet, www.orpha.net,(ultimo accesso 12.10.2023)
- Di Lazzaro V, et al: Niemann-Pick di tipo C: focus sulla forma ad esordio adolescenziale/adulto. Int J Neurosci 2016; 126(11): 963-971.
- Hammerschmidt TG, et al: Biomarcatori molecolari e biochimici per la diagnosi e il monitoraggio della terapia nei pazienti affetti da Niemann-Pick di tipo C. Int J Dev Neurosci 2017; 66: 18-23.
- Bonnot O, et al.: Sintomi psichiatrici e neurologici nei pazienti con malattia di Niemann-Pick di tipo C (NP-C): Risultati del Registro Internazionale NPC. World J Biol Psychiatry 2017: 1-10.
- “Malattia di Niemann-Pick”, https://flexikon.doccheck.com,(ultimo accesso 12.10.2023)
- Desnick JP, et al.: Identificazione e caratterizzazione di otto nuove mutazioni SMPD1 che causano la malattia di Niemann Pick di tipo A e B. Mol Med 2010; 16: 316-321.
- “Xenpozyme® (olipudase alfa) approvato dalla Commissione Europea come primo e unico trattamento per l’ASMD”, 28.06.2022.
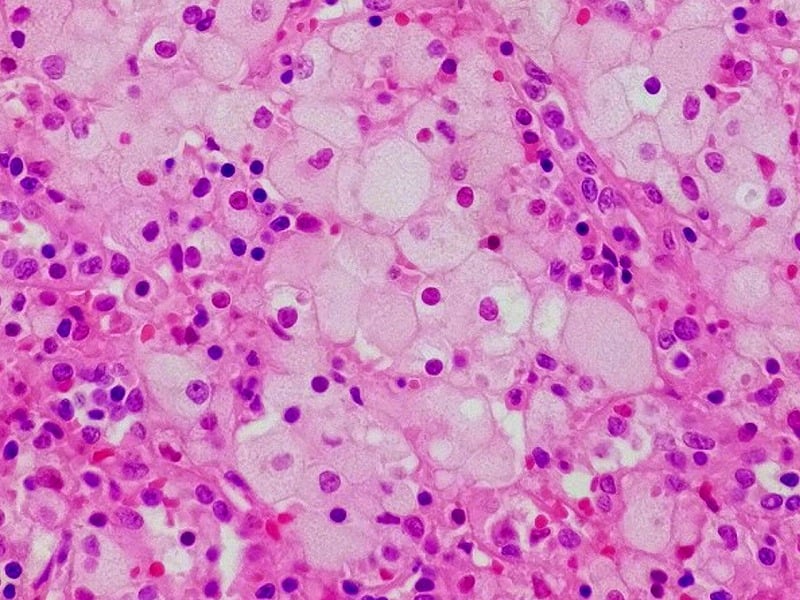
| Immagine di copertina: cellula Niemann pick nella milza. ©W.CC, Wikimedia |
PRATICA GP 2023; 18(10): 48