
L’aspettativa di vita e la qualità della vita dei pazienti affetti da SLA possono essere migliorate con una terapia moderna. La volontà del paziente è fondamentale e deve essere sempre determinata di nuovo. Non ci sono nuovi sviluppi nella terapia farmacologica – il riluzolo deve essere iniziato presto. All’inizio della malattia, è consigliabile una valutazione dettagliata nell’ambiente ospedaliero di una clinica neurologica. Si raccomanda un ulteriore trattamento in centri specializzati durante il decorso della malattia.
Nella sclerosi laterale amiotrofica (SLA), la più comune malattia dei motoneuroni, si verifica una perdita progressiva di cellule nervose nel sistema motorio. Questo colpisce in genere sia i primi motoneuroni del tratto piramidale che i secondi motoneuroni delle cellule del corno anteriore. Allo stesso tempo, la gravità può variare, soprattutto all’inizio della malattia, e i segni del primo o del secondo motoneurone possono dominare. Lo spettro delle malattie del motoneurone comprende altre patologie come la sclerosi laterale primaria (PLS) o l’atrofia muscolare spinale (SMA), che a loro volta colpiscono solo il primo o il secondo arto. secondo motoneurone.
L’incidenza della SLA è rara rispetto ad altre malattie. Tuttavia, l’incidenza è ancora di circa 2/100.000 abitanti ed è quindi solo leggermente inferiore all’incidenza della sclerosi multipla, per esempio. Al contrario, la prevalenza è molto bassa, pari a 3-8/100.000 abitanti [1]. Questo riflette indirettamente la breve sopravvivenza media dei pazienti dopo la diagnosi, che nella maggior parte dei casi è di soli due o quattro anni. È interessante notare, tuttavia, che circa il 10% dei pazienti ha un decorso molto più lento, con una sopravvivenza superiore ai dieci anni.
La terapia di supporto moderna e massima può oggi prolungare significativamente la sopravvivenza dei pazienti e migliorare la loro qualità di vita, almeno per quanto riguarda i sintomi essenziali come il dolore, la fame e la mancanza di respiro. Tuttavia, soprattutto alla luce delle migliori possibilità mediche, è molto importante mettere sempre i desideri della persona colpita al centro delle decisioni terapeutiche [2]. A tal fine, è necessario redigere precocemente un testamento biologico dettagliato, che dovrà essere rivisto più volte con il progredire della malattia.
Principi di diagnostica
I capisaldi della diagnostica della SLA continuano ad essere l’anamnesi e l’esame clinico. Se questi vengono eseguiti da un neurologo esperto, di solito è già evidente la presenza di un danno progressivo al primo e/o al secondo motoneurone. Solo questo può essere utilizzato per diagnosticare la SLA probabile o certa, secondo i criteri diagnostici validi [3]. I risultati clinici possono essere ulteriormente corroborati dai risultati elettrofisiologici, che ora sono sempre più inclusi nell’algoritmo diagnostico (Fig. 1) [4].
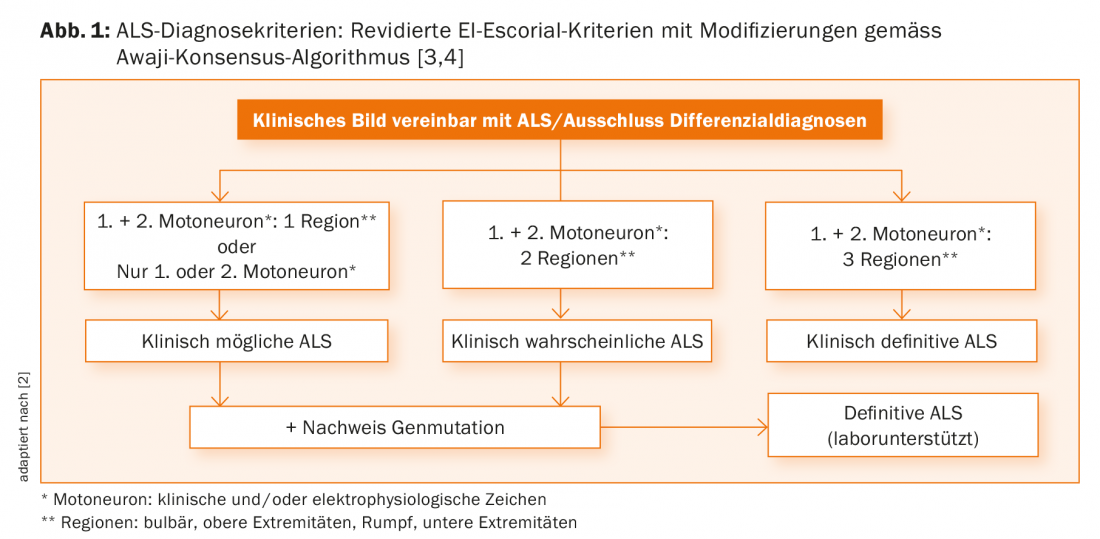
Purtroppo, con i criteri attuali, spesso una diagnosi affidabile può essere fatta solo in ritardo nel corso della malattia, il che, da un lato, può turbare i medici di riferimento, i pazienti e i parenti, e dall’altro non fornisce una buona base per gli studi scientifici. Pertanto, la revisione dei criteri diagnostici continua ad essere di grande importanza.
Un compito diagnostico essenziale è quello di escludere qualsiasi diagnosi differenziale che possa causare una costellazione di sintomi analoga. Queste sono chiaramente riassunte nelle attuali linee guida EFNS [2]. Ad esempio, la combinazione non così rara di stenosi spinale cervicale e polineuropatia potrebbe fornire la combinazione di segni del primo e del secondo motoneurone necessari per la diagnosi di SLA. Una valutazione in regime di ricovero in una clinica neurologica all’esordio della malattia si è dimostrata efficace per fornire spazio e tempo sufficienti per la valutazione clinica, l’esclusione delle diagnosi differenziali e la comunicazione empatica della diagnosi.
Esame clinico
L’esame clinico serve innanzitutto a cercare segni di danno al primo e al secondo motoneurone nelle quattro regioni del corpo (bulbare, arti superiori, tronco, arti inferiori). I segni del primo motoneurone includono spasticità, clonia, iperreflessia e fenomeni di disinibizione centrale. I segni clinici del secondo motoneurone sono principalmente fascicolazioni e atrofie. Inoltre, si devono ricercare segni atipici nell’esame clinico.
Ad esempio, un coinvolgimento dei muscoli oculari o anche una mancanza di progressione indirizzano l’attenzione verso diagnosi alternative. I disturbi sensoriali, d’altra parte, non escludono completamente la SLA, ma anch’essi devono certamente essere motivo di un’intensa chiarificazione delle diagnosi differenziali.
Elettrofisiologia
L’elettroneurografia (ENG) può essere utilizzata per escludere la polineuropatia. Nell’elettromiografia (EMG), è tipica della SLA una costellazione di sintomi composta da segni di danno acuti, subacuti e forse cronici, che riflettono il decorso della malattia e la capacità conservata degli assoni periferici di rigenerarsi. Oltre alla ricerca di attività spontanee patologiche sul muscolo a riposo, si presta particolare attenzione alla stabilità e alle dimensioni delle unità motorie nell’analisi del potenziale singolo. Un riscontro patologico corrispondente nell’EMG è una parte essenziale della diagnosi di SLA e può essere considerato uguale a un segno clinico secondo i criteri diagnostici modificati [4].
I potenziali evocati motori possono rilevare un danno subclinico al tratto piramidale. I metodi di quantificazione delle unità motorie (stima del numero di unità motorie) possono essere descritti come metodi piuttosto scientifici. Questi metodi potrebbero diventare sempre più importanti, soprattutto per la progressione della malattia e quindi anche per gli studi clinici [5].
Imaging
L’importanza degli ultrasuoni nella diagnosi della SLA è aumentata negli ultimi anni, anche se non si prevede che il metodo raggiunga l’importanza che ha nelle malattie dei nervi periferici. L’ecografia nervosa può essere particolarmente utile per escludere diagnosi differenziali rilevanti, come le neuropatie immuno-mediate. In particolare, gli ultrasuoni possono essere utilizzati anche per esaminare i muscoli. Per il rilevamento delle fascicolazioni, l’ecografia muscolare, con la sua eccellente sensibilità e una specificità leggermente inferiore, potrebbe essere in grado di sostituire almeno parzialmente l’elettromiografia in futuro, soprattutto per gli esami di follow-up [6]. La risonanza magnetica è particolarmente importante per escludere le diagnosi differenziali. Nella maggior parte dei casi, vengono visualizzati il cervello e l’intera colonna vertebrale. Inoltre, possono essere imitati anche i muscoli e i nervi periferici, anche se l’attenzione in questo caso è ancora più nel campo scientifico che nella pratica clinica.
bugie.
Diagnostica genetica
La diagnostica genetica nella SLA è stata ampliata da diversi aspetti negli ultimi anni. Da molti anni si cercano in particolare mutazioni nel gene della superossido dismutasi Cu/Zn SOD1, che sono responsabili di circa il 10-15% dei casi di SLA familiare. Paradossalmente, ci sono famiglie in cui il rilevamento della mutazione non è correlato all’insorgenza della malattia, il che indica che devono esistere altri fattori o geni che causano la malattia [7]. Nel frattempo, è stato trovato un nuovo locus genetico essenziale a questo proposito, il gene C9orf72. Le mutazioni in questo gene sono responsabili di circa il 25% dei casi familiari e di circa il 10% dei casi sporadici. È interessante notare che le mutazioni in questo gene sono presenti anche in una percentuale simile di casi di demenza frontotemporale, evidenziando il legame tra le due malattie anche a livello genetico [8].
I test genetici delle mutazioni più comuni sono oggi disponibili in commercio. È quindi ancora più importante sottolineare che la diagnostica genetica deve essere affidata a mani esperte, soprattutto nel caso della SLA. Dovrebbe essere ordinato solo dopo un esame e una consulenza approfonditi da parte di neurologi e genetisti umani che conoscono la malattia, in quanto il risultato può avere una grande rilevanza, soprattutto per la consulenza ai familiari asintomatici.
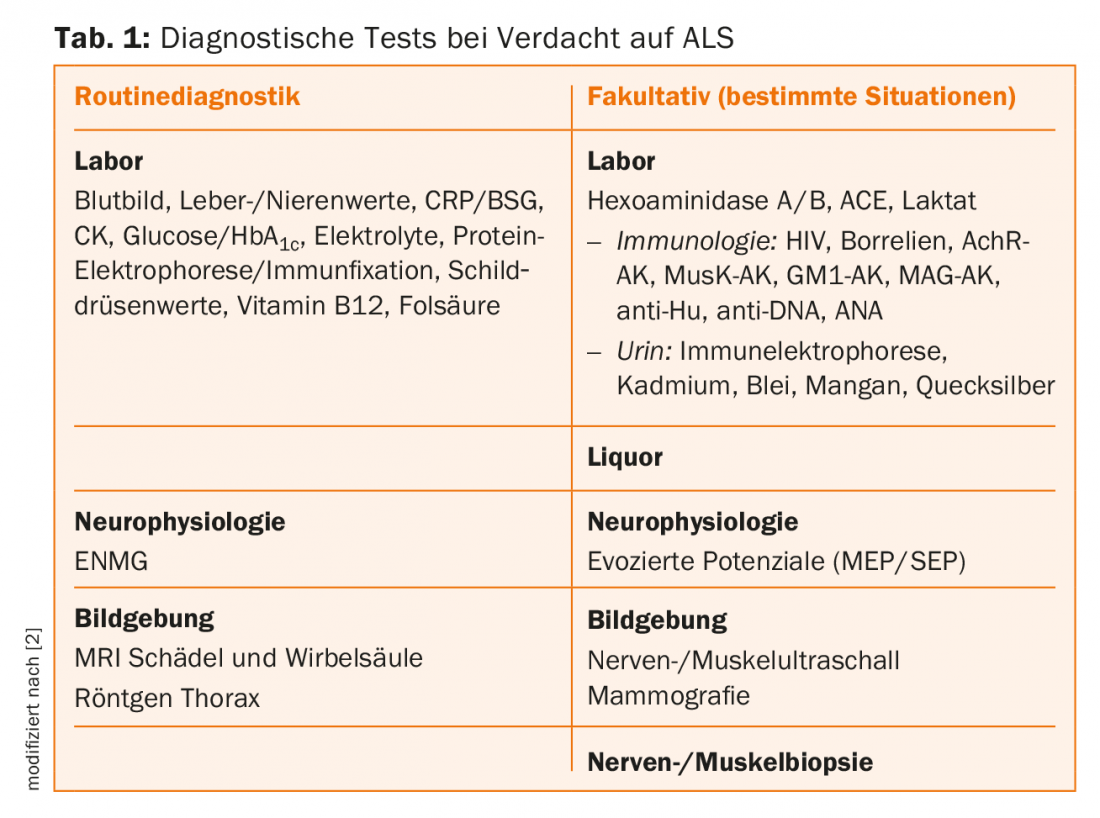
Ulteriori diagnosi
Se si sospettano anomalie cognitive, si raccomanda un test neuropsicologico. A seconda della durata della malattia, fino al 50% dei pazienti affetti da SLA sviluppa sintomi disesecutivi e in circa il 15% può essere fatta una diagnosi di demenza fronto-temporale. Questa osservazione contraddice le dottrine precedenti che non vedevano alcun danno cognitivo nella SLA. Tra l’altro, i pazienti con demenza fronto-temporale dovrebbero anche essere esaminati molto attentamente per individuare i segni della malattia del motoneurone, che si verifica più frequentemente nel corso della malattia. Altrimenti, ulteriori diagnosi servono essenzialmente a escludere le diagnosi differenziali [2].
Terapia farmacologica
Inoltre, il riluzolo è l’unico farmaco approvato per il trattamento della SLA; si raccomanda di prescriverlo il più presto possibile nel corso della malattia. Il farmaco è sicuro; oltre a un lieve affaticamento, raramente si verifica un aumento degli enzimi epatici. Il decorso della malattia può essere rallentato in qualche modo con il riluzolo. Inoltre, possono essere utilizzate altre terapie farmacologiche orientate ai sintomi, come l’amitriptilina o le gocce di atropina per disturbare la salivazione. I farmaci potenzialmente sedativi, come le benzodiazepine e gli oppiacei, possono essere utilizzati molto bene per trattare l’ansia, il dolore e la dispnea, entro limiti terapeutici relativamente ristretti.
Nutrizione
La perdita di peso è un marcatore prognostico negativo che dovrebbe essere evitato, se possibile. I pazienti e i loro parenti possono essere consigliati di conseguenza e si possono offrire integratori nutrizionali. Un attento monitoraggio logopedico per il rilevamento precoce della disfagia è di grande importanza, in modo che l’offerta di nutrizione enterale percutanea mediante un tubo PEG possa essere fatta il prima possibile. Quanto più precocemente nel corso della malattia viene inserita la PEG, tanto minore è il rischio di complicazioni e tanto più probabile è che la misura possa contribuire a migliorare la qualità della vita e il tempo di sopravvivenza. Importante per le persone colpite è il fatto che l’assunzione di altri alimenti per via orale rimane possibile come ovvio.
Ventilazione
In qualsiasi momento del decorso della malattia, faccia attenzione ai sintomi di ipoventilazione, soprattutto di notte. Oltre alla dispnea, questi includono la sonnolenza diurna, il mal di testa mattutino o l’ortopnea. Questi sintomi possono essere trattati in modo molto efficace con la ventilazione domiciliare non invasiva, che migliora significativamente la qualità di vita delle persone colpite. Questo efficace trattamento sintomatico dovrebbe essere discusso con tutte le persone colpite in una fase iniziale, quando compaiono i sintomi corrispondenti, in quanto può migliorare la qualità della vita delle persone colpite, almeno per un periodo limitato, come quasi nessun’altra terapia [9]. La ventilazione invasiva, invece, viene presa in considerazione solo per un gruppo selezionato di pazienti. Se a un certo punto del decorso della malattia il paziente desidera interrompere la ventilazione, questo deve essere rispettato e accompagnato di conseguenza. Di norma, questa misura porta presto allaritenzione di CO2 e i pazienti muoiono in un’anestesia daCO2 a lenta insorgenza.
Ulteriore terapia
Un trattamento specializzato e multiprofessionale, come quello fornito in un centro SLA o in istituzioni analoghe, è essenziale per una cura ottimale dei pazienti affetti da SLA. Da un lato, le terapie sopra citate possono essere applicate in una fase precoce e coordinate in modo ottimale; dall’altro, possono essere utilmente integrate altre misure terapeutiche come la fisioterapia, la terapia occupazionale e la logopedia, che mirano a preservare le funzioni rimanenti, come la capacità di comunicare, il più a lungo possibile.
Letteratura:
- Schweikert K: Sclerosi laterale amiotrofica. Swiss Medical Forum 2015; 15: 1068-1073.
- Andersen PM, et al: Linee guida EFNS sulla gestione clinica della sclerosi laterale amiotrofica (MALS) – relazione rivista di una task force EFNS. Eur J Neurol 2012; 19: 360-375.
- Brooks BR, et al: El Escorial rivisitato: criteri rivisti per la diagnosi di sclerosi laterale amiotrofica. Sclerosi Laterale Amiotrofica Altri Disordini del Motoneurone 2000; 1: 293-299.
- Carvalho MD, et al: L’algoritmo diagnostico Awaji aumenta la sensibilità dei criteri El Escorial per la diagnosi di SLA. Sclerosi Laterale Amiotrofica 2009; 10: 53-57.
- Schulte-Mattler WJ, et al: MUNIX – un promettente biomarcatore nella SLA. Clin Neurophysiol 2015; 46: 186-189.
- Schreiber S, et al: Stato dell’esperienza e significato dei metodi di imaging nei cambiamenti neuromuscolari indotti dalla SLA. Clin Neurophysiol 2016; 46: 173-181.
- Felbecker A, et al.: Quattro pedigree familiari di SLA discordanti per due mutazioni SOD1: tutte le mutazioni SOD1 sono patogene? J Neurol Neurosurg Psychiatry 2010; 81: 572-577.
- Rohrer JD, et al: Espansioni di C9orf72 nella demenza frontotemporale e nella sclerosi laterale amiotrofica. Lancet Neurol 2015; 14: 291-301.
- Boentert M, et al: Gestione della ventilazione e della secrezione nella sclerosi laterale amiotrofica. Clin Neurophysiol 2015; 46: 163-172.
InFo NEUROLOGIA & PSICHIATRIA 2016; 14(2): 26-29









