L’amiloidosi è una rara forma di dermatosi da deposito causata dall’errato ripiegamento delle proteine. La diagnosi precoce dermatologica è di grande importanza per l’ulteriore decorso della malattia.
Etimologicamente, il termine amiloidosi risale al greco antico ἄμυλον ámylon, che significa “pasto energetico, amido”. Poiché biochimicamente è presente una struttura proteica, si tratta di un nome fuorviante [1]. Gli amiloidi sono un gruppo di proteine eterogenee che appaiono simili ultrastrutturalmente o reagiscono in modo simile nella colorazione istologica. Le amiloidosi sia localizzate che sistemiche possono essere caratterizzate da sintomi cutanei. “Più di 25 proteine sono in grado di formare l’amiloide nelle persone sane”, ha detto il Dr. med. Antonio Cozzio, Clinica di Dermatologia, Venereologia e Allergologia dell’Ospedale Cantonale di San Gallo, che ha parlato di questo argomento alle Giornate di Formazione Dermatologica di Zurigo di quest’anno. Delle proteine biochimicamente diverse che sono in grado di formare l’amiloide, 15 possono portare a malattie clinicamente rilevanti [2].

Cambiare la tassonomia
La diagnosi precoce da parte del dermatologo è una base importante per un trattamento efficace e per la prevenzione di un ulteriore deterioramento degli organi coinvolti [3]. Nel 2016, il sistema di classificazione della Società per le amiloidosi è stato rivisto [4]: La congofilia e la birifrangenza sono ancora i criteri classificatori più importanti. Inoltre, si raccomanda di determinare l’identità chimica della proteina della fibrilla amiloide depositata nello spazio extracellulare dei tessuti mediante l’analisi della sequenza proteica. Ad oggi, sono note 36 pro- teine fibrillari extracellulari nell’uomo, 2 delle quali sono di natura iatrogena, 9 sono state identificate anche negli animali. Due proteine fibrillari scoperte di recente sono AApoCII (derivato dell’apolipoproteina CII) e AApoCIII (derivato dell’apolipoproteina CIII). Le amiloidosi AApoCII e AApoCIII sono amiloidosi sistemiche ereditarie. Due proteine precedentemente considerate inclusioni intracellulari, la tau e l’α-sinucleina, sono ora classificate come depositi extracellulari associati alla morte cellulare, denominati “ATau” e “AαSyn”.
Coinvolgimento cutaneo eterogeneo
Si distingue tra forme sistemiche e cutanee limitate di amiloidosi, ciascuna delle quali è suddivisa in sottotipi acquisiti ed ereditari (Fig. 1).
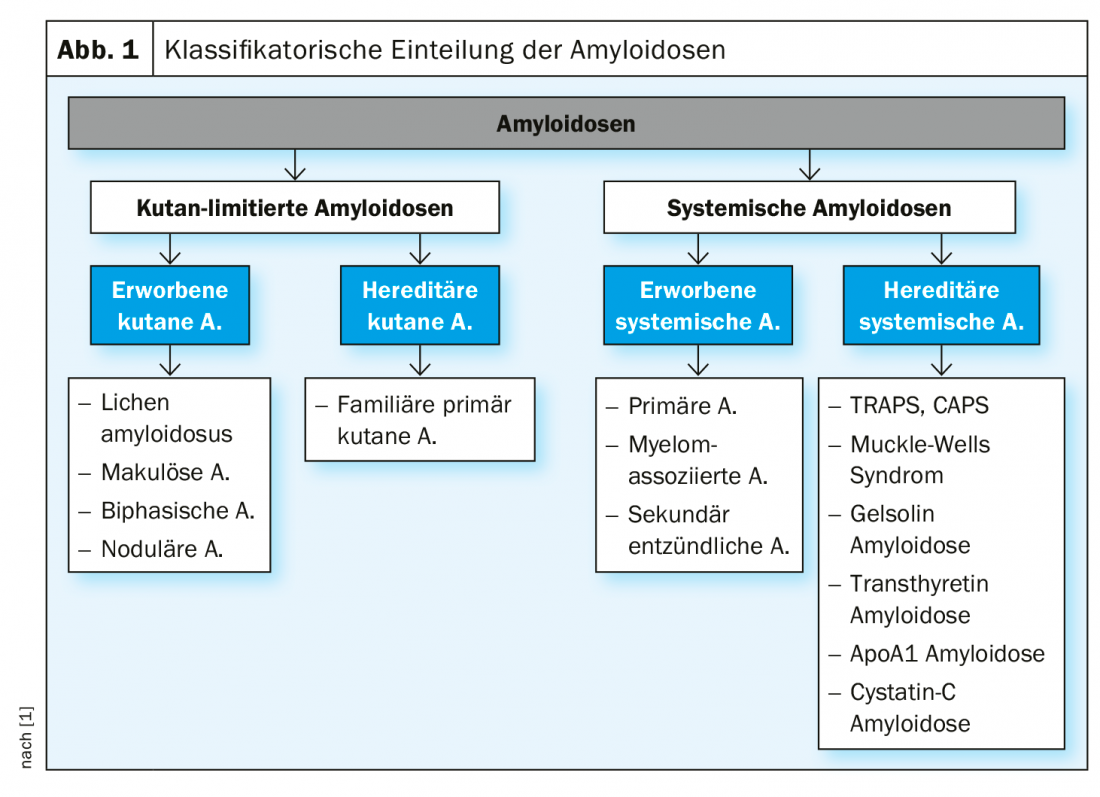
Amiloidosi sistemiche: si tratta di un quadro clinico eterogeneo, caratterizzato dalla deposizione di proteine dell’organismo modificate in modo patologico nei tessuti o negli organi. I segni cutanei caratteristici delle diverse forme di amiloidosi sistemica acquisita sono riassunti nella Tabella 1.
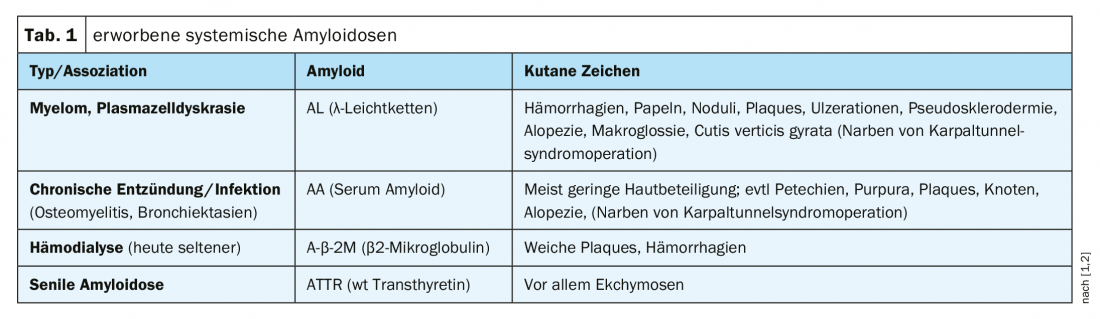
L’amiloidosi a catena leggera è la forma più comune e spesso è molto aggressiva. Se vengono colpiti il cuore o i reni, questa forma di amiloidosi può portare alla morte dopo pochi mesi, se non viene trattata. Si ritiene che le catene leggere anticorpali svolgano un ruolo importante nell’eziologia e portino ai caratteristici depositi nel tessuto. L’amiloidosi sistemica è comune nei pazienti con mieloma multiplo. In una fase iniziale dell’amiloidosi sistemica primaria, le manifestazioni cliniche sono inizialmente limitate a sintomi dermatologici; il coinvolgimento degli organi si verifica solo nel decorso successivo [5]. Lo spettro diagnostico comprende la biopsia del tessuto e del midollo osseo, l’elettroforesi e gli esami specifici dell’organo (panoramica 1).


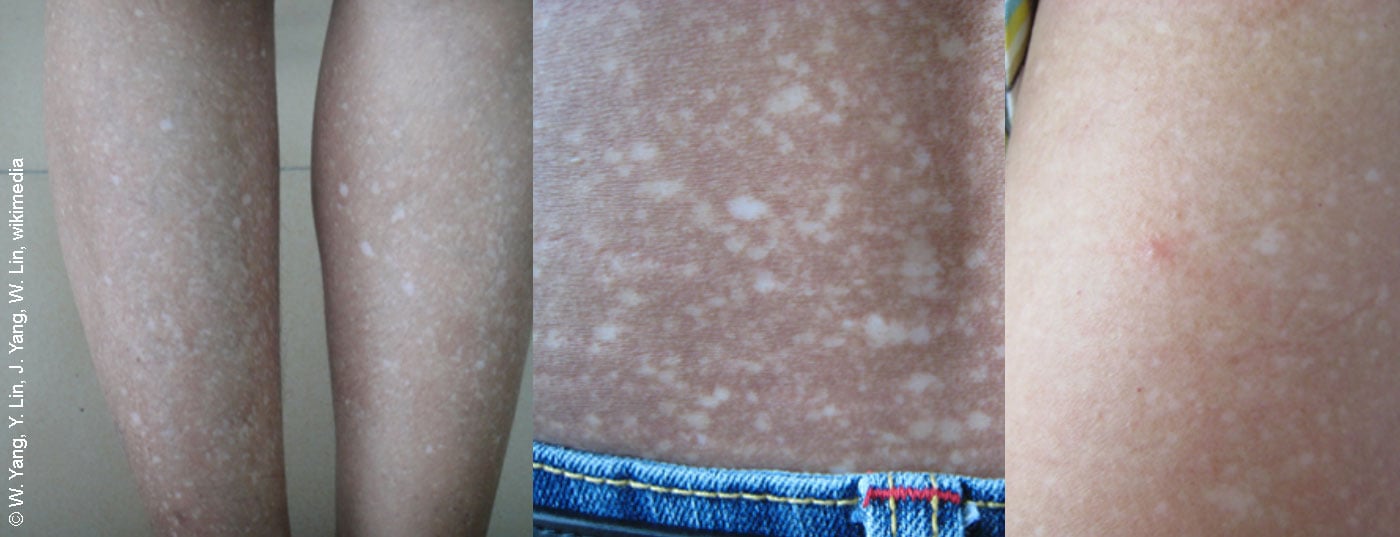
Amiloidosi cutanee limitate: questo termine collettivo descrive le patologie dermatologiche che sono istologicamente caratterizzate dall’accumulo extracellulare di depositi amiloidi nel derma. “Le amiloidosi maculari e papulari sono solitamente limitate a livello cutaneo”, spiega il relatore. Se si sospetta un’amiloidosi cutanea nodale, occorre chiarire se è presente un coinvolgimento cardiaco/renale; può essere necessaria la collaborazione con l’ematooncologia [1]. In caso di porpora non chiara (soprattutto in caso di formazione di noduli aggiuntivi, macroglossia), si raccomanda una biopsia con analisi immunoistochimica, in cui possono essere inclusi anche i risultati istologici dell’anamnesi precedente. Devono essere escluse le seguenti diagnosi differenziali: neoplasia cutanea, sarcoidosi, granuloma faciale, leishmaniosi (panoramica 2) [1]. Lo spettro delle opzioni di trattamento è relativamente ampio e deve essere adattato individualmente (riquadro).
Letteratura:
- Cozzio A: Presentazione diapositive: Tema annuale delle dermatosi deposizionali. Amiloidosi, Antonio Cozzio, MD, 9° Giornate di formazione in dermatologia di Zurigo 2019, Zurigo, 26 giugno 2019.
- Rauch PJ: Amiloidosi sistemiche. Switzerland Med Forum 2014; 14(50): 943-948.
- Bruch-Gerharz D, Ruzicka T: Amiloidosi e ialinosi. In: Plewig G., Ruzicka T., Kaufmann R, Hertl M (eds) Braun-Falco’s Dermatology, Venereology and Allergology 2017. Springer Reference Medicine. Berlino, Heidelberg: Springer.
- Sipe JD, et al: Proteine a fibrilla amiloide e amiloidosi: identificazione chimica e classificazione clinica Linee guida della Società Internazionale di Amiloidosi 2016. Amiloide. Il Journal of Protein Folding Disorders (Giornale dei disturbi del ripiegamento delle proteine)
- Trajber Horvat A, Trčko K, Jurčić V, Marko PB: Amiloidosi sistemica primaria con coinvolgimento cutaneo e cardiaco: un caso clinico. Acta Dermatovenerologica Alpina, Pannonica, et Adriatica 2018. www.oneamyloidosisvoice.com/rcuratenew
DERMATOLOGIE PRAXIS 2019; 29(5): 27-28 (pubblicato il 10.10.19, prima della stampa).