Se una o entrambe le camere del cuore sono eccessivamente ispessite o dilatate, il cuore non è più efficiente. Le cardiomiopatie sono caratterizzate da una varietà di cause diverse di tali cambiamenti nel tessuto muscolare cardiaco. Tuttavia, la differenziazione delle singole manifestazioni cliniche ha importanti implicazioni per la prognosi e i possibili regimi di trattamento.
Le cardiomiopatie hanno molti volti. Fondamentalmente, possono essere classificate in quattro fenotipi morfologici: cardiomiopatia ipertrofica (HCM), cardiomiopatia dilatativa (DCM), cardiomiopatia aritmogena (ARVC) e cardiomiopatia restrittiva (RCM). Tuttavia, questa classificazione approssimativa, basata sulla visione, ha poco in comune con le cause reali, come si può vedere dal gran numero di sottocategorie. Il Prof. Dr. med. Benjamin Meder, Heidelberg (D), ha dimostrato che la sola diagnostica per immagini non è quindi sufficiente per rilevare la malattia. Di conseguenza, occorre aggiungere le caratteristiche funzionali. Questo perché la cardiomiopatia è in definitiva la descrizione di un fenotipo morfologico e funzionale del miocardio che non può essere spiegato dalla malattia coronarica o dalle proprietà di riempimento alterate dovute all’ipertensione arteriosa, alla viziatura valvolare o alla cardiopatia congenita.
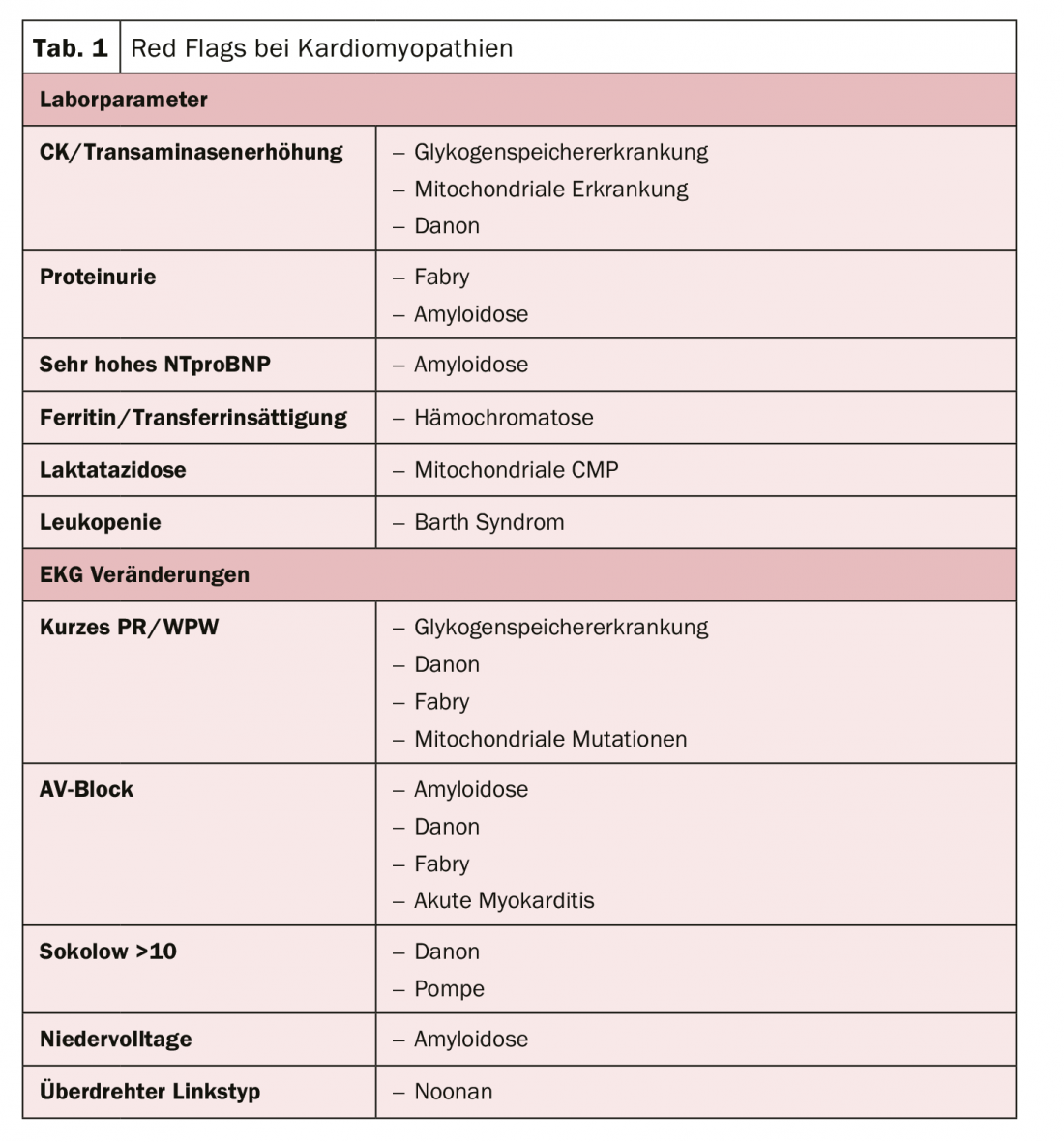
L’approccio strutturale di un trattamento ottimizzato passa quindi attraverso l’eziologia, con l’obiettivo di evitare la morte cardiaca improvvisa, il problema limitante di tutte le cardiomiopatie. Ci sono parametri di laboratorio tipici e anomalie nell’ECG che possono indicare la presenza di una cardiomiopatia (tab. 1). Essere in grado di fare queste distinzioni è quindi importante, poiché la prognosi è essenzialmente determinata dalla causa, dice l’esperto. Soprattutto i pazienti con amiloidosi hanno una sopravvivenza molto scarsa.
Individuare l’amiloidosi e trattarla in modo efficace
L’amiloidosi può essere suddivisa in tre tipi in base al coinvolgimento degli organi: L’amiloidosi AL colpisce i reni, il cuore e/o l’intestino, l’amiloidosi mt-ATTR colpisce soprattutto il cuore o il sistema nervoso, e l’amiloidosi wt-ATTR colpisce soprattutto il cuore. In linea di principio, l’amiloidosi AL può essere intesa come una complicazione della discrasia plasmacellulare. Pertanto, viene utilizzata una terapia ematologica simile a quella del mieloma multiplo, con l’obiettivo di eliminare le catene leggere amiloidogeniche e (cardio-)tossiche il più rapidamente possibile. Oltre alla chemioterapia ad alte dosi per i pazienti idonei, viene utilizzato principalmente un regime basato sugli inibitori del proteasoma. Nei pazienti più giovani, CyBorD (bortezomib, ciclofosfamide, desametasone) dovrebbe essere preferito a BMDex (bortezomib, melfalan, desametasone) a causa della tossicità per le cellule staminali del melfalan, al fine di preservare l’opzione di una successiva aferesi delle cellule staminali e della chemioterapia ad alte dosi.
Nell’amiloidosi causata da depositi di proteina transtiretina, i farmaci come diflunisal e tafamidis possono stabilizzare la proteina mutata. Inoltre, le terapie geniche che riducono la produzione di transtiretina (ad esempio, patisiran, inotersen) possono ridurre gli effetti sul sistema nervoso.
Per i pazienti con AL cardiaca sintomatica e amiloidosi ATTR, si applicano in linea di massima le stesse raccomandazioni terapeutiche generali dei pazienti con insufficienza cardiaca. Tuttavia, anche basse dosi di β-bloccanti o ACE-inibitori possono provocare ipotensione sintomatica. Pertanto, il trattamento nei pazienti con amiloidosi cardiaca si basa principalmente sul corretto dosaggio dei diuretici.
Coinvolgimento sindromico di molti sistemi di organi – malattia di Fabry
La malattia di Fabry è un disturbo geneticamente ereditato che di solito si manifesta tra i 20 e i 40 anni. Si forma un enzima α-Gal A mal ripiegato o non funzionale, per cui il trasporto dal reticolo endoplasmatico al lisosoma è disturbato. Questo porta ad un accumulo di substrati lisosomiali. Oltre al cuore, ai reni, al sistema nervoso centrale, possono essere coinvolti anche il sistema nervoso periferico e la pelle. Se non viene trattata, l’insufficienza renale è la causa più frequente di morte in questi pazienti, ha spiegato la Prof. Dr med. Ingrid Kindermann, Homburg/Saar (D). I sintomi cardiaci si manifestano in più della metà dei pazienti Fabry. Le aritmie maligne sono le principali responsabili della morte cardiaca improvvisa.
Per molto tempo, la terapia si è limitata ad alleviare i sintomi. Nel frattempo, esiste un’opzione di trattamento specifica in cui l’enzima mancante viene sostituito da un enzima prodotto biotecnologicamente. L’alfa-galattosidasi A prodotta in laboratorio viene somministrata al paziente tramite infusione e garantisce la rottura del materiale di accumulo. Tuttavia, poiché la terapia enzimatica sostitutiva non può riparare i danni agli organi che si sono già verificati, ma solo ritardarne la progressione, dovrebbe essere utilizzata il prima possibile. Un’altra opzione è quella di assumere un chaperon farmacologico per via orale. Questo agente può essere utilizzato nei pazienti con determinate mutazioni nel gene GLA e con un’attività residua dell’enzima α-galattosidasi A. Questo perché si lega alle forme instabili di AGAL e stabilizza l’enzima in modo che possa scomporre le sostanze grasse accumulate nella cellula.
Fonte: CardioMedLive 2020
CARDIOVASC 2020; 19(3): 28-29 (pubblicato il 17/9/20, prima della stampa).