Un trattamento precoce e adeguato può avere un effetto positivo sull’ulteriore decorso di questa rara malattia multisistemica. Questo articolo offre una breve panoramica di ciò che si conosce oggi. Tra l’altro, l’identificazione di nuovi biomarcatori potrebbe servire come base per una terapia personalizzata in futuro.
La dermatomiosite giovanile (JDM) è una malattia sistemica, immuno-mediata dell’infanzia, caratterizzata da debolezza muscolare e tipiche eruzioni cutanee [9]. Anche altri sistemi di organi, come il cuore e i polmoni, possono essere colpiti e sono ancora la causa più comune di morte in questi pazienti. Si tratta della miopatia infiammatoria idiopatica più comune nell’infanzia, con un’incidenza di 2-4 milioni all’anno.
Il processo infiammatorio è caratterizzato dall’infiltrazione del tessuto colpito da parte delle cellule del sistema immunitario, come le cellule T e le cellule dendritiche plasmacitoidi, e da una firma di interferone [1]. La maggior parte dei sintomi è dovuta alla vasculopatia, che comporta una mancanza di funzionamento e una perdita di cellule endoteliali. Un’immunosoppressione efficace, di solito una terapia combinata di metotrexato sottocutaneo e corticosteroidi orali, ha ridotto notevolmente la mortalità (attualmente circa l’1%) e circa il 50% dei pazienti presenta una sola riacutizzazione della malattia prima di raggiungere la remissione [2]. Il restante 50% presenta un decorso cronico, con frequenti ricadute, aumento della morbilità e riduzione della qualità di vita [2].

Sottofenotipizzazione attraverso i biomarcatori
La JDM è una malattia molto eterogenea con diversi fenotipi, per cui diversi sistemi di organi possono essere colpiti in misura variabile. Gli autoanticorpi sono stati oggetto di un’intensa ricerca negli ultimi anni e sono stati in grado di caratterizzare meglio i diversi fenotipi; pertanto, gli autoanticorpi hanno un valore prognostico [3]. Il monitoraggio dell’attività della malattia e del danno agli organi causato dall’infiammazione è una parte importante della gestione di questi giovani pazienti, poiché determina l’esito a lungo termine di questa malattia [4]. Il medico ha a disposizione diversi strumenti per farlo, come l’istopatologia del tessuto muscolare e cutaneo, la microscopia capillare (Fig. 1), i punteggi clinici per valutare il coinvolgimento muscolare e cutaneo [5] e ora anche diversi biomarcatori nel sangue [6,7]). I punteggi clinici utilizzati per valutare l’attività della malattia richiedono la collaborazione del paziente, che a volte può essere impegnativa nei bambini più piccoli. Questi punteggi clinici sono anche di utilità limitata nel rilevare l’infiammazione sottostante o nel differenziare altre cause di debolezza muscolare, come la tossicità da steroidi.

Linee guida per ottimizzare la gestione della malattia
Nel 2012 è stata lanciata l’iniziativa europea SHARE (Single Hub and Access point for paediatric Rheumatology in Europe) con l’obiettivo di standardizzare la gestione diagnostica e terapeutica dei bambini e degli adolescenti con malattie reumatiche in tutta Europa. Per il JDM, queste raccomandazioni sono state pubblicate nel 2017 [6].
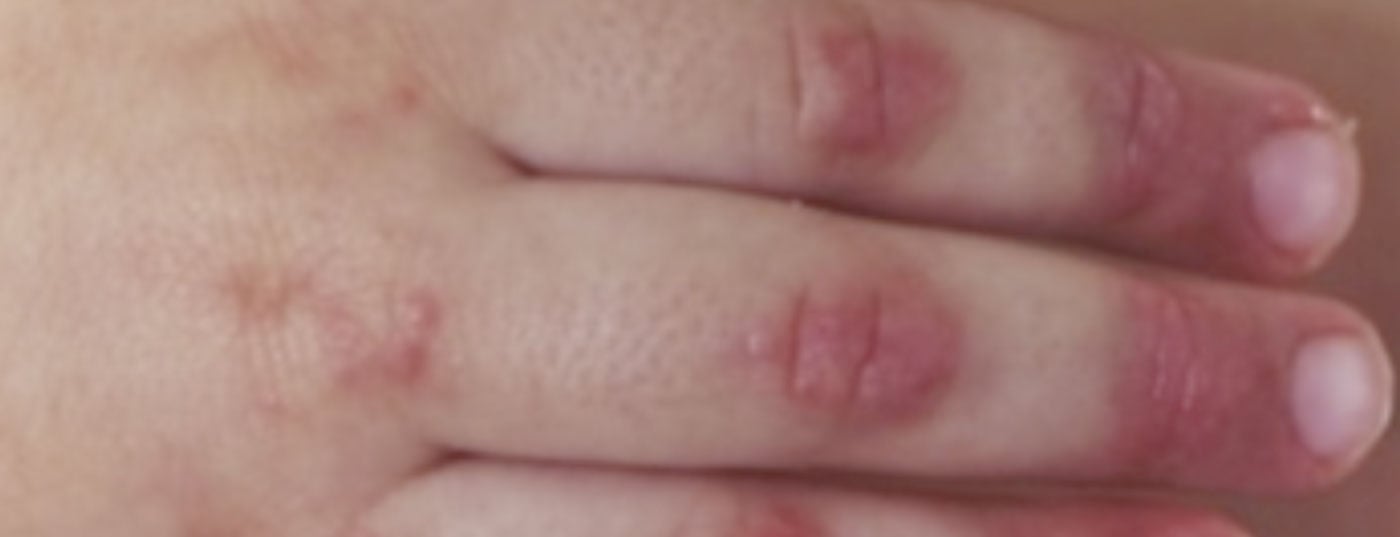
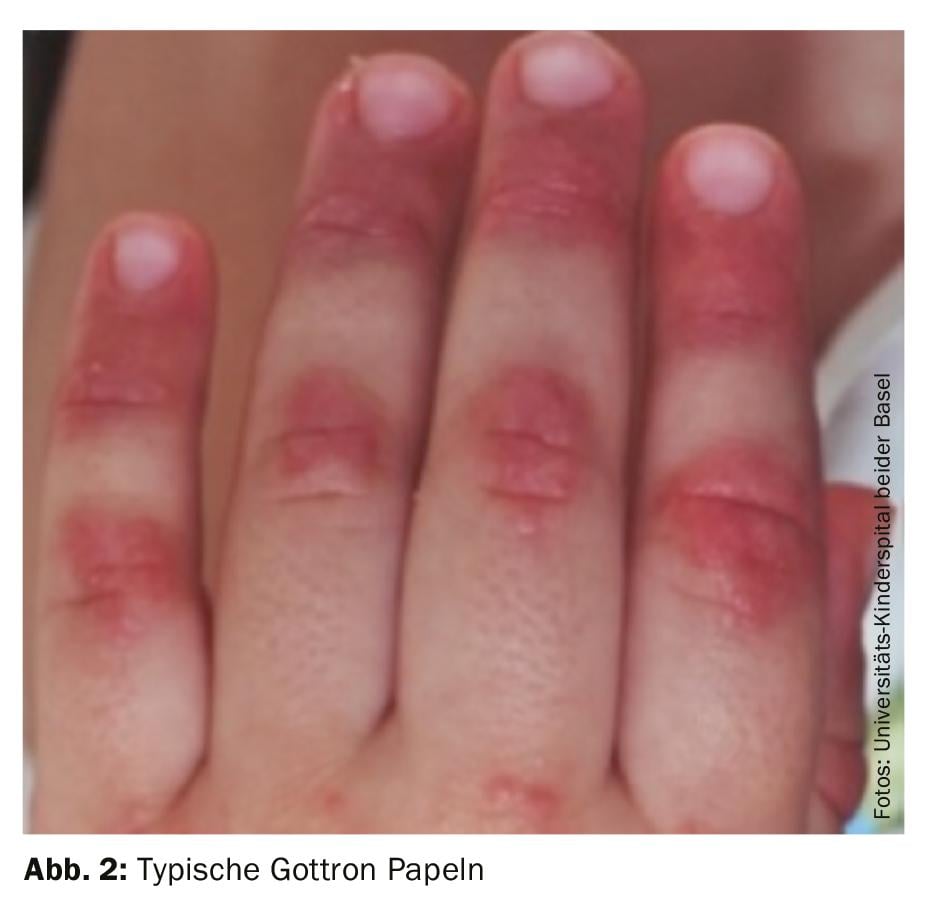
Diagnosi: la diagnosi di dermatomiosite giovanile rimane principalmente clinica, con i tipici reperti cutanei (Fig. 2) e la debolezza muscolare evidenziata prossimalmente. Pertanto, in caso di sospetto di JDM, si deve fare un rapido rinvio a un centro di eccellenza vicino. Una terapia precoce e adeguata può prevenire i danni a lungo termine e aumentare le possibilità di remissione precoce. I criteri diagnostici classici di Bohan e Peter del 1975 contengono cinque elementi:
- segni cutanei caratteristici
- Debolezza muscolare prossimale
- tipica biopsia muscolare patologica
- enzimi muscolari elevati
- alterazioni miopatiche nell’elettromiogramma (EMG)
→ Nella pratica reumatologica pediatrica, l’EMG è stato sostituito dalla risonanza magnetica non dolorosa (Fig. 3) [8].

Nel 2017 sono stati pubblicati nuovi criteri di classificazione che, ad esempio, omettono la biopsia muscolare per i sintomi cutanei patognomici e includono anche gli autoanticorpi (anti-Jo-1) nei criteri [9]. Anche gli ultrasuoni sono sempre più utilizzati, perché non richiedono la sedazione nei bambini più piccoli.
La debolezza muscolare deve essere valutata nel bambino con diversi strumenti. Sono disponibili test muscolari funzionali, il cosiddetto Children’s Myositis Activity Score (CMAS) [10] e test muscolari che misurano solo la forza, il cosiddetto Manual Muscle Test (MMT) [11]. Entrambi i test sono utili per valutare la debolezza muscolare al momento della diagnosi; ma servono anche come importanti parametri di follow-up che possono dare un’indicazione significativa dell’attività della malattia. Entrambi i test fanno parte dei criteri di remissione del 2013 [12].
Wienke et al. hanno recentemente convalidato diversi biomarcatori (galectina-9 e CXCL10) che svolgono un ruolo non solo nella diagnosi, ma anche nel follow-up e possono anche facilitare le decisioni terapeutiche all’inizio della malattia: In questo studio prospettico, i pazienti con livelli elevati di questi biomarcatori al momento della diagnosi hanno mostrato un decorso più grave nel follow-up e quindi potrebbero potenzialmente beneficiare di una terapia più intensiva. Al contrario, nei pazienti con una sola ricaduta, i valori si erano quasi normalizzati già alcuni mesi dopo la diagnosi, sotto terapia. Inoltre, è stato possibile dimostrare che i marcatori mostravano già un nuovo aumento nel sangue prima dei risultati clinici di una recrudescenza della malattia [7].

Terapia 2: la terapia di prima linea rimane una combinazione di steroidi e me-trexato, eventualmente ciclosporina [6]. Il metotrexato deve essere mantenuto per almeno 2 anni; gli steroidi vengono sospesi nel 1° anno di terapia nel caso di un ciclo monofasico [6]. Se alcuni sintomi, come una grave debolezza muscolare (il bambino non può più alzarsi dal letto, disfagia grave), la compromissione della funzione miocardica o polmonare o le ulcere cutanee indicano un decorso grave, è necessario aggiungere la ciclofosfamide. Se non ci sono miglioramenti, si devono aggiungere immunoglobuline per via endovenosa o ciclosporina A, oppure passare ai biologici (rituximab, infliximab, adalimumab). Da un punto di vista pediatrico, l’aumento della calcinosi del tessuto sottocutaneo o muscolare (Figg. 3 e 4) è un segno di infiammazione sistemica persistente, che giustifica l’intensificazione della terapia. I trapianti di cellule staminali sono stati eseguiti con successo anche nei pazienti con JDM con un decorso molto grave; alcuni pazienti hanno raggiunto la remissione completa senza terapia. Quando si raggiunge la remissione clinica, la calcinosi può risolversi completamente.
A fronte di un buon successo, questa terapia standard rimane associata al rischio di effetti collaterali indesiderati, come le infezioni, la debolezza muscolare indotta dagli steroidi o la nausea settimanale con il metotrexato. In futuro, i nuovi biomarcatori citati potrebbero consentire l’identificazione precoce di una nuova recrudescenza della malattia o una più rapida riduzione della terapia. I biomarcatori potrebbero quindi supportare il percorso verso una terapia personalizzata per questa malattia rara.
| Strumento diagnostico basato sul web Nel frattempo, è disponibile uno strumento web che, oltre alla diagnosi, fornisce anche una probabilità della diagnosi con la classificazione del sottogruppo. Questi criteri sono stati convalidati sia per i bambini che per gli adulti con sospetta miopatia infiammatoria. http://www.imm.ki.se/biostatistics/calculators/iim/ |
Letteratura:
- Piper CJM, et al: Le cellule B CD19(+)CD24(hi)CD38(hi) sono espanse nella dermatomiosite giovanile e presentano un fenotipo pro-infiammatorio dopo l’attivazione attraverso il Toll-Like Receptor 7 e l’interferone-alfa. Front Immunol 2018; 9: 1372.
- Gowdie PJ, Allen RC, Kornberg AJ, Akikusa JD: Caratteristiche cliniche e decorso della malattia nei pazienti con dermatomiosite giovanile. Int J Rheum Dis 2013; 16(5): 561-567.
- Rider LG, Nistala K: Le miopatie infiammatorie idiopatiche giovanili: patogenesi, fenotipi clinici e autoanticorpali ed esiti. J Intern Med 2016; 280(1): 24-38.
- Huber A, Feldman BM: Esiti a lungo termine nella dermatomiosite giovanile: come siamo arrivati e dove stiamo andando? Curr Rheumatol Rep 2005; 7(6): 441-446.
- Huber AM, et al: Convalida preliminare e significato clinico del Cutaneous Assessment Tool nella dermatomiosite giovanile. Arthritis Rheum 2008; 59(2): 214-221.
- Enders FB, et al: Raccomandazioni basate sul consenso per la gestione della dermatomiosite giovanile. Ann Rheum Dis 2017; 76(2): 329-340.
- Wienke J, et al: Galectina-9 e CXCL10 come biomarcatori dell’attività della malattia nella dermatomiosite giovanile: uno studio di coorte longitudinale e una convalida multi-corte. Arthritis Rheumatol 2019. doi.org/10.1002/art.40881
- Brown VE, et al: Un’indagine di consenso internazionale sui criteri diagnostici della dermatomiosite giovanile (JDM). Reumatologia (Oxford) 2006; 45(8): 990-993.
- Lundberg IE, et al: Criteri di classificazione 2017 della European League Against Rheumatism/American College of Rheumatology per le miopatie infiammatorie idiopatiche adulte e giovanili e i loro principali sottogruppi. Ann Rheum Dis 2017; 76(12): 1955-1964.
- Huber AM, et al: Validazione e significato clinico della Childhood Myositis Assessment Scale per la valutazione della funzione muscolare nelle miopatie infiammatorie idiopatiche giovanili. Arthritis Rheum 2004; 50(5): 1595-1603.
- Jain M, et al: Affidabilità intra-rater e inter-rater del Manual Muscle Test (MMT) di forza a 10 punti nei bambini con miopatie infiammatorie idiopatiche giovanili (JIIM). Phys Occup Ther Pediatr 2006; 26(3): 5-17.
- Lazarevic D, et al: I criteri PRINTO per la malattia clinicamente inattiva nella dermatomiosite giovanile. Ann Rheum Dis 2013; 72(5): 686-693.
- Mathiesen PR, et al: Fitness aerobico dopo il JDM – uno studio di follow-up a lungo termine. Rheumatology (Oxford) 2013; 52(2): 287-295.
PRATICA DERMATOLOGICA 2019; 29(4): 26-29