Il trattamento di scelta per il sarcoma di Ewing comprende la chemioterapia neoadiuvante, la resezione del tumore e la chemioterapia post-operatoria con/senza radioterapia. In questo contesto, la gestione interdisciplinare presso un centro appropriato è di importanza cruciale.
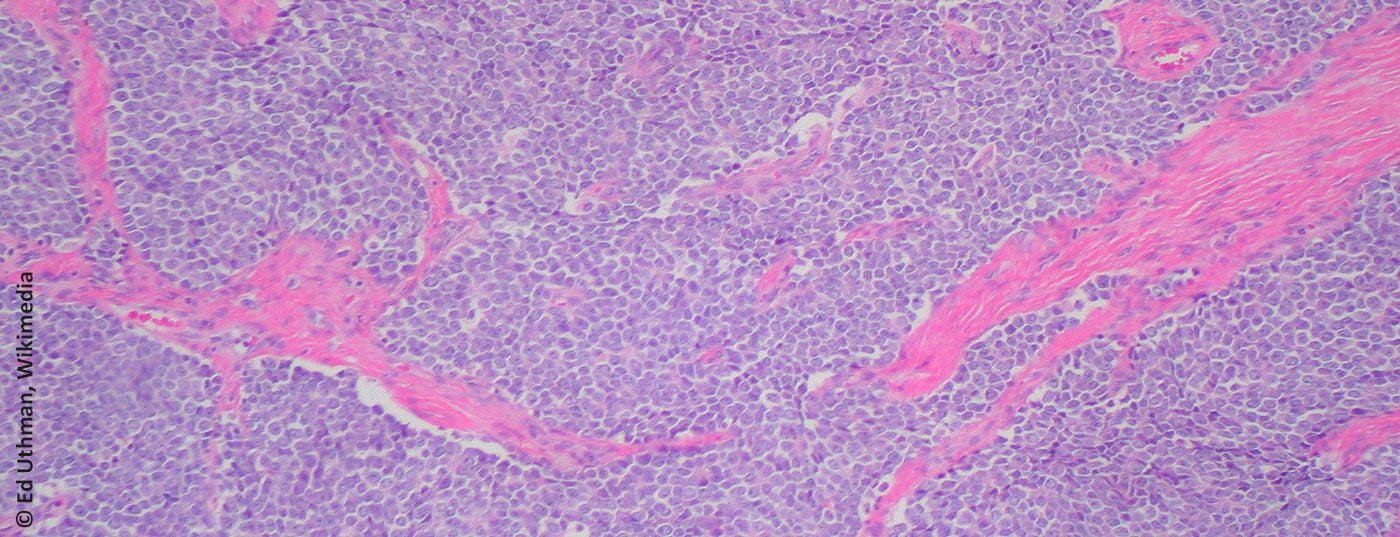
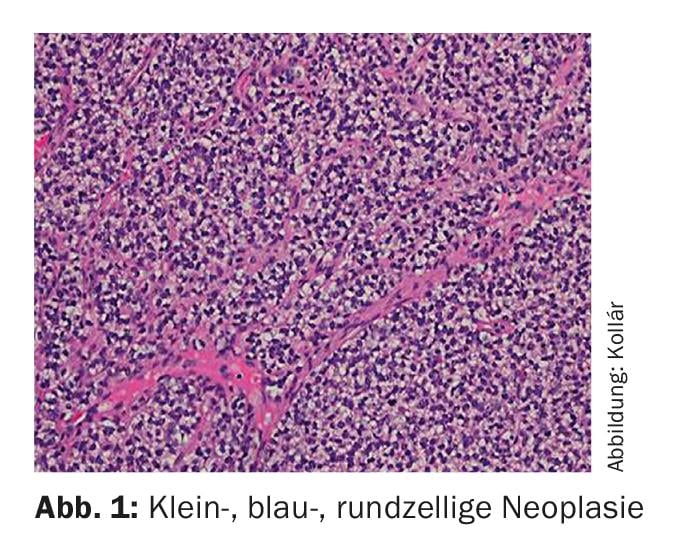
Il sarcoma di Ewing prende il nome da James Ewing (patologo americano, 1866-1943). Dal punto di vista microscopico, appartiene al gruppo dei piccoli tumori a cellule rotonde di colore blu (Fig. 1) . La cellula di origine non è chiaramente compresa, anche se la presenza di marcatori neuronali suggerisce un collegamento con il neuroectoderma embrionale [1]. La diagnosi viene solitamente effettuata mediante il rilevamento diagnostico molecolare di traslocazioni che coinvolgono il gene EWS sul cromosoma 22. La traslocazione più comune (85-95%) è t(11;22)(q24;q12) [2]. Per definizione, tutti i sarcomi di Ewing sono classificati come altamente maligni (G3) [3].
Epidemiologia
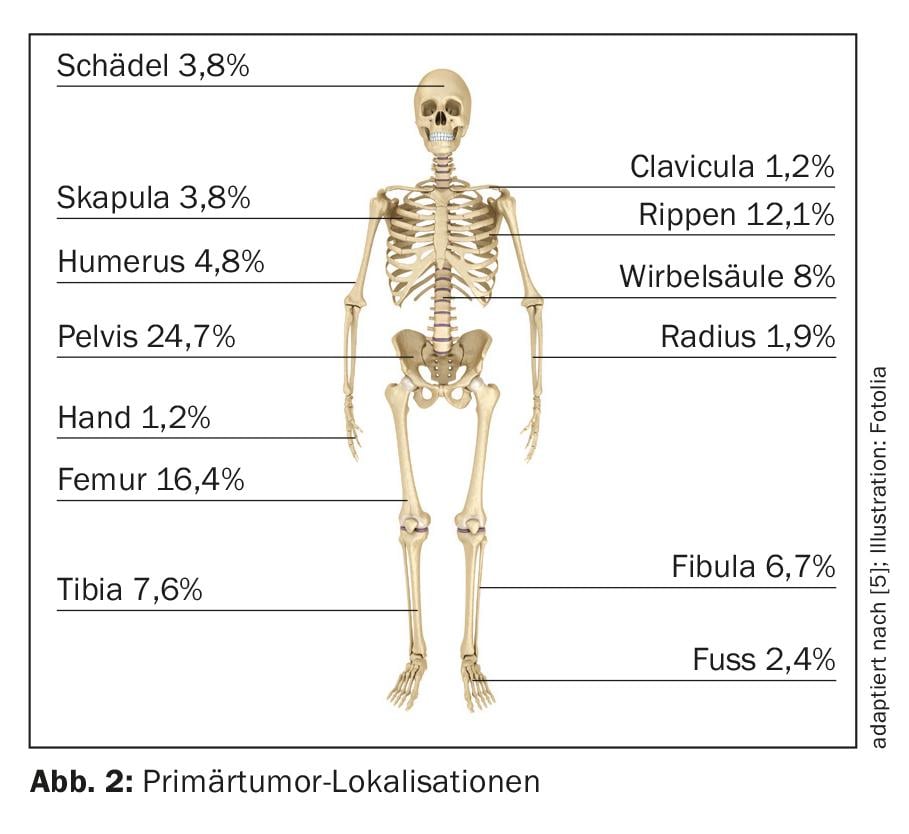
Dopo l’osteosarcoma, il sarcoma di Ewing rappresenta il secondo tumore osseo maligno primario più comune nell’infanzia e nell’adolescenza. L’età mediana di insorgenza è di 10-15 anni, il tasso di incidenza annuale è di circa 3/1’000’000 di popolazione [4]. Il sesso maschile è colpito leggermente più spesso (1,5:1). Il bacino (25%) e le diafisi delle ossa tubolari lunghe, soprattutto nel femore (circa 16% ) (Fig. 2), sono tra le localizzazioni primarie più frequenti del tumore [5]. Nel 15% dei casi, si può documentare principalmente una localizzazione extraossea [6].
Clinica
I sintomi iniziali sono aspecifici. Un dolore localizzato e/o un gonfiore, possibilmente con conseguente restrizione della mobilità, sono in primo piano tra i sintomi. In circa il 10-15% dei casi, alla diagnosi iniziale è presente una frattura patologica [7], nell’80% uno stadio tumorale formalmente localizzato. A causa di un tasso di metastasi molto elevato (>80%) dopo una terapia esclusivamente locale del tumore primario, si può presumere che in quasi tutti i casi siano già presenti metastasi subcliniche [8]. Le metastasi si verificano più frequentemente a livello polmonare, osseo e nel midollo osseo [9].
Diagnostica
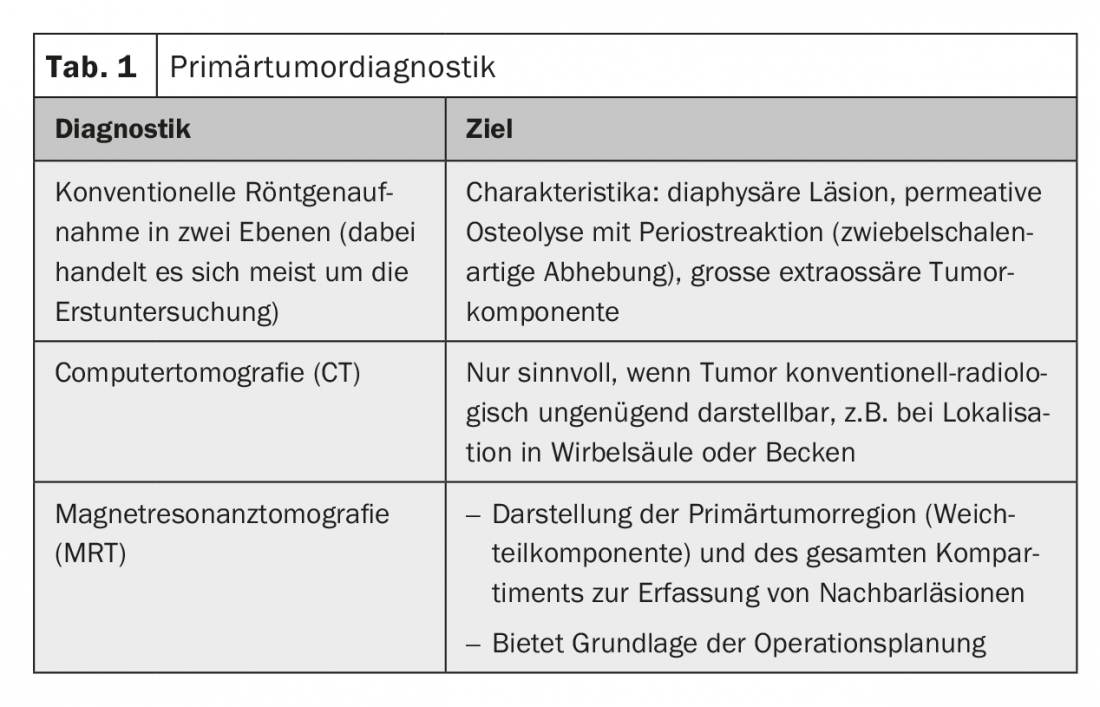
Diagnosi radiologica del tumore primario: la diagnosi radiologica del tumore primario deve sempre essere effettuata prima della biopsia. È la base per valutare l’importanza del tumore e la resecabilità, nonché per pianificare la biopsia (tab. 1).
Biopsia: se si sospetta un tumore maligno dei tessuti molli o dell’osso, la biopsia è obbligatoria per confermare la diagnosi. La biopsia deve essere eseguita con il coinvolgimento di un chirurgo esperto nel trattamento dei sarcomi, idealmente il futuro chirurgo. A questo proposito, è necessario prendere in considerazione il percorso di accesso chirurgico. Il gold standard è la biopsia ad ago centrale.
La messa in scena: Dopo la conferma della diagnosi con la biopsia, si devono organizzare i seguenti esami di stadiazione:
- TC torace (esclusione di metastasi polmonari)
- Scintigrafia scheletrica (esclusione di metastasi ossee)
- Biopsia e aspirazione del midollo osseo (indicata solo se la PET/CT non viene eseguita a causa della bassa incidenza).
- Ulteriori procedure di imaging a seconda dei disturbi clinici.
Il valore di un esame PET/CT nella stadiazione iniziale e come imaging di follow-up è attualmente oggetto di studi clinici. La sensibilità del rilevamento delle metastasi polmonari è inferiore rispetto alla TAC del torace, mentre la sensibilità del rilevamento delle lesioni ossee è superiore [10,11].
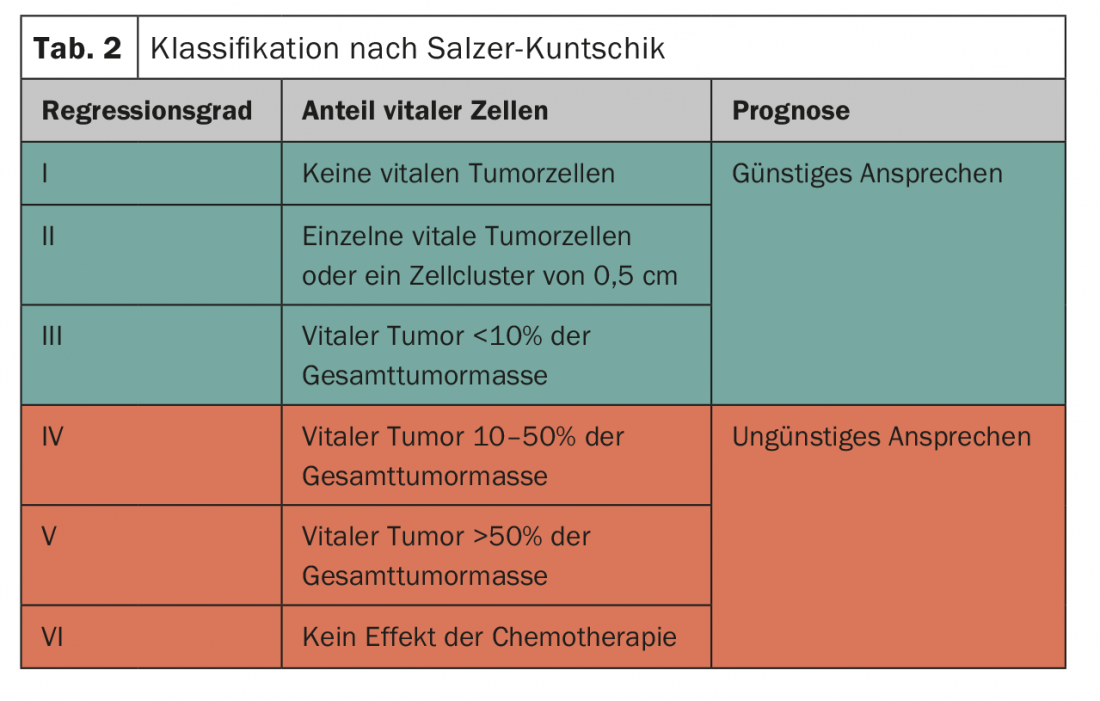
Risposta istologica del tumore alla terapia di sistema: per valutare la risposta alla chemioterapia, si determina la percentuale di cellule maligne vitali nel campione di resezione definitivo. Il patologo classifica poi la risposta nei Paesi di lingua tedesca in base a questa valutazione (Tab. 2) [12]. La classificazione ha un significato prognostico; è di grande importanza soprattutto nel contesto degli studi Euro-Ewing per determinare la strategia terapeutica post-operatoria.
Stadio localizzato della malattia
La terapia del sarcoma di Ewing è multimodale e deve essere discussa in modo interdisciplinare in un centro di sarcomi. Questo include sempre la terapia locale (resezione e/o radioterapia) e la chemioterapia. I primi studi prospettici randomizzati hanno dimostrato una sopravvivenza globale significativamente migliore con la chemioterapia aggiuntiva (10-20% contro poco meno del 70% nella sopravvivenza libera da eventi a 5 anni) [8,13], motivo per cui l’attuale standard di cura comprende la chemioterapia neoadiuvante seguita dalla terapia locale del tumore primario e dalla chemioterapia adiuvante [14]. I pazienti devono essere trattati nell’ambito di un protocollo di studio, se possibile.
Chirurgia: la resezione del tumore primario è la terapia locale di scelta. L’obiettivo è sempre quello di ottenere una resezione completa del tumore. Tuttavia, la resecabilità del tumore primario a volte non viene indicata a seconda della localizzazione anatomica. Questi includono principalmente i sarcomi di Ewing della colonna vertebrale e del bacino. In questi casi, la modalità di terapia deve essere personalizzata. In questo caso, la resezione con radioterapia post-operatoria o la radioterapia da sola sono mirate a [15,16].
Radioterapia: in quanto tumori sensibili alle radiazioni, i sarcomi di Ewing mostrano un tasso di controllo locale comparabile con la radioterapia rispetto ai pazienti trattati chirurgicamente in alcuni studi [17]. La radioterapia definitiva è preferita per i tumori primari non resecabili secondo una resezione incompleta seguita da radioterapia post-operatoria [17]. In caso di escissione marginale/intralesionale, c’è l’indicazione di eseguire la radioterapia post-operatoria. Il valore della radioterapia additiva nei casi di risposta istologica insufficiente alla chemioterapia (ma con resezione completa del tumore) non è chiaro. In linea di principio, la radioterapia può essere somministrata anche in fase preoperatoria [18].
Terapia di sistema: lo standard internazionale è la chemioterapia di combinazione. Gli agenti chemioterapici più efficaci includono le sostanze alchilanti (ifosfamide, ciclofosfamide), le antracicline (doxorubicina), nonché etoposide, vincristina e actinomicina. Il regime VIDE (vincristina, ifosfamide, doxorubicina, etoposide) in analogia al protocollo Euro-Ewing 1999 e 2008 in Europa e il regime VDC/IE (agenti VIDE + ciclofosfamide) in America servono spesso come modelli per la chemioterapia di induzione. L’aggiunta di ifosfamide ed etoposide alla VDC è stata associata a un significativo prolungamento della sopravvivenza libera da eventi a 5 anni nello studio randomizzato IESS-III (69% vs. 54%) [19]. Il confronto diretto tra gli schemi viene testato nel più recente studio Euro-Ewing 2012.
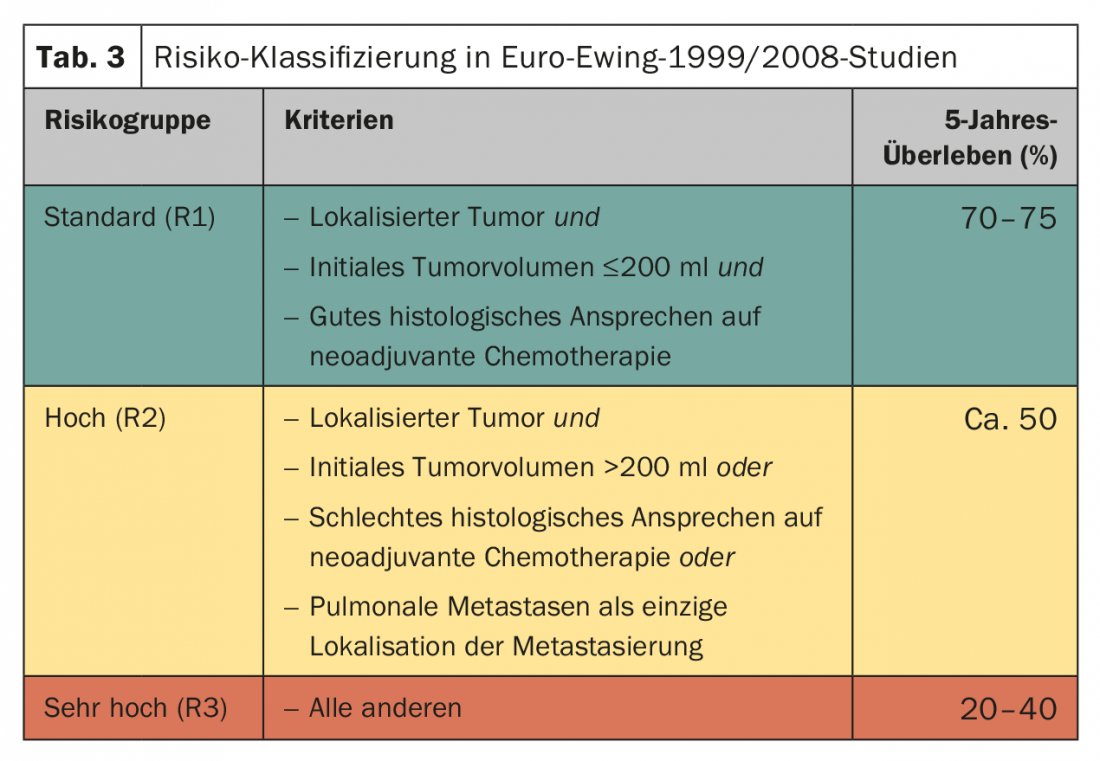
La terapia locale di solito segue sei cicli di chemioterapia neoadiuvante. La terapia post-operatoria viene pianificata in base alla costellazione del rischio (tab. 3). Lo standard è di otto cicli di chemioterapia secondo VAI o VAC. Lo studio Euro-Ewing 1999 ha confrontato in modo casuale l’efficacia di VAC (ciclofosfamide) e VAI (ifosfamide) nel gruppo di rischio R1. Fondamentalmente, è stata confermata l’equivalenza dei regimi, con il sesso maschile che sembra beneficiare maggiormente della VAI (HR 1,34; 95% CI 0,96-1,86) [20]. La durata totale della terapia è di circa dieci-dodici mesi. Nel gruppo di rischio R2 (stadio del tumore localizzato, scarsa risposta istologica del tumore, volume del tumore >200 ml), è stata randomizzata l’importanza della terapia ad alte dosi contenente melfalan con trapianto di cellule staminali autologhe. In questo caso, un miglioramento significativo della sopravvivenza globale è stato ottenuto con la chemioterapia ad alte dosi con busulfano/melfalan (77,8 vs 69,9%, HR 0,60 [0,39–0,92], p=0,019) [21].
Stadio della malattia metastatica
Nello stadio metastatizzato, il sarcoma di Ewing risponde fondamentalmente agli stessi agenti chemioterapici utilizzati nello stadio localizzato. A seconda del numero e soprattutto della localizzazione, esiste un potenziale curativo nonostante la presenza di metastasi ematogene. Per le metastasi polmonari/ pleuriche isolate, il tasso di guarigione è fino al 40%, per le metastasi ossee e del midollo osseo è di circa il 20-25% e per una combinazione di queste sedi è del 15% [22]. Questi risultati si ottengono in particolare con una terapia locale coerente di tutte le metastasi.
Metastasi polmonari/pleuriche: Se le metastasi polmonari persistenti vengono resecate dopo la chemioterapia di induzione, questo sembra essere associato a un miglioramento della prognosi [23] – corrispondente alla raccomandazione di rimuovere chirurgicamente le metastasi polmonari radiologicamente visibili durante il decorso. Poiché spesso si vedono più focolai intraoperatori rispetto a quelli oggettivati dalla stadiazione preoperatoria, la resezione dovrebbe essere eseguita con la chirurgia aperta [24].
Secondo analisi retrospettive, l’irradiazione parenchimale del polmone (tra 15 e 20 Gy) è anche associata a una prognosi più favorevole nei casi di metastasi isolate pneumo-pleuriche [25,26]. L’indicazione deve quindi essere valutata nella remissione completa dopo la chemioterapia e dopo la resezione di tutti i focolai polmonari.
Metastasi ossee e del midollo osseo: In caso di metastasi ossee e/o del midollo osseo, la prognosi è scarsa nonostante l’obiettivo terapeutico potenzialmente curativo. In caso di coinvolgimento oligometastatico, si raccomanda di considerare le opzioni di terapia locale. La radioterapia è il metodo principale utilizzato in questo caso.
Chemioterapia ad alto dosaggio con trapianto di cellule staminali autologhe: il valore della chemioterapia ad alto dosaggio con supporto di cellule staminali autologhe è stato a lungo discusso in letteratura. Uno studio prospettico non randomizzato ha mostrato risultati eccezionali, con una sopravvivenza libera da eventi a 5 anni fino al 52%. Tuttavia, altre pubblicazioni non hanno potuto supportare questi risultati [27,28]. Solo recentemente, i risultati della stratificazione R2 sono stati riportati nel contesto di Euro-Ewing 1999. Nei pazienti con metastasi polmonari, la chemioterapia ad alte dosi senza irradiazione polmonare non ha dimostrato un beneficio rispetto alla chemioterapia convenzionale con irradiazione polmonare [29]. I risultati del gruppo di rischio R3 sono ancora in sospeso.
Trattamento delle recidive
Le recidive più frequenti si verificano nei primi cinque anni dopo la diagnosi iniziale, ma anche le recidive tardive non sono rare [30]. La prognosi delle recidive entro i primi due anni è molto scarsa, mentre le recidive successive mostrano una sopravvivenza a lungo termine in circa il 15-20% [31]. La terapia di scelta dipende dal momento della recidiva, dalla localizzazione e dal numero di manifestazioni tumorali e dalla terapia precedente. Le recidive locali e le metastasi polmonari isolate vengono solitamente trattate localmente, cioè con la resezione e/o la radioterapia [32]. In caso di focolai estesi di recidiva, è nuovamente indicato l’avvio di una terapia sistemica, anche se non esiste un regime standard per questo. Se c’è una risposta buona e, soprattutto, lunga alla terapia iniziale, si può valutare una ripetizione di questa. La dose cumulativa di doxorubicina non deve essere trascurata. C’è una tendenza generale a intensificare la chemioterapia con una terapia ad alte dosi, anche se le prove a questo proposito sono limitate [33]. I regimi chemioterapici utilizzati includono topotecan/ciclofosfamide, irinotecan/temozolomide, gemcitabina/docetaxel, infosfamide ad alto dosaggio e chemioterapia contenente platino con etoposide [34–36]. Le terapie a bersaglio molecolare (ad esempio, gli inibitori di IGF-1 e PARP) e gli approcci immunoterapeutici sono attualmente oggetto di sperimentazione.
Cura successiva
Il follow-up mira a individuare precocemente le recidive e a monitorare le tossicità tardive. Attualmente, mancano dati prospettici che dimostrino un beneficio in termini di sopravvivenza derivante da esami di follow-up regolari. Gli intervalli di follow-up sono un tentativo di rendere giustizia alla maggiore probabilità di recidiva nei primi due o tre anni. Le raccomandazioni si trovano, ad esempio, nelle linee guida del National Comprehensive Cancer Network (NCCN).
Le più comuni sono le neoplasie secondarie (incidenza cumulativa 9%), le endocrinopatie, tra cui. Infertilità, cardio-, nefro- e neurotossicità, tossicità polmonare e compromissione funzionale locoregionale nel contesto della terapia locale come tossicità terapeutiche tardive su [37]. I dettagli e le raccomandazioni sul monitoraggio degli effetti tardivi sono disponibili su www.survivorshipguidelines.org.
Messaggi da portare a casa
- La diagnosi e la terapia del sarcoma di Ewing vengono effettuate in modo interdisciplinare in un centro di sarcomi. La diagnosi radiologica del tumore primario deve essere effettuata prima della biopsia. Il trattamento di scelta è quello neoadiuvante.
- Chemioterapia, resezione del tumore e chemioterapia post-operatoria con/senza radioterapia.
- Il tumore deve essere resecato da un chirurgo esperto in sarcomoncologia.
- Anche nello stadio di tumore metastatizzato, l’obiettivo terapeutico è curativo.
Letteratura:
- Lipinski M, et al: Antigeni associati al neuroectoderma su linee cellulari di sarcoma di Ewing. Ricerca sul cancro 1987; 47: 183-187.
- de Alava E, Gerald WL: Biologia molecolare della famiglia del sarcoma di Ewing/ tumore neuroectodermico primitivo. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2000; 18: 204-213.
- Fletcher CDM BJ, Hogendoorn P, Mertens F (eds.): Classificazione OMS dei tumori dei tessuti molli e delle ossa (IARC WHO Classification of Tumours). 4° ed. 2013.
- Esiashvili N, Goodman M, Marcus RB Jr: Cambiamenti nell’incidenza e nella sopravvivenza dei pazienti affetti da sarcoma di Ewing negli ultimi 3 decenni: dati della Surveillance Epidemiology and End Results. Journal of pediatric hematology/oncology 2008; 30: 425-430.
- Cotterill SJ, et al: Fattori prognostici nel tumore di Ewing dell’osso: analisi di 975 pazienti dello European Intergroup Cooperative Ewing’s Sarcoma Study Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2000; 18: 3108-3114.
- Applebaum MA, et al: Caratteristiche cliniche ed esiti nei pazienti con sarcoma di Ewing extrascheletrico. Cancro 2011; 117: 3027-3032.
- Widhe B, Widhe T: Sintomi iniziali e caratteristiche cliniche nell’osteosarcoma e nel sarcoma di Ewing. Il Journal of bone and joint surgery volume americano 2000; 82: 667-674.
- Nesbit ME Jr, et al: Terapia multimodale per la gestione del sarcoma di Ewing primario, non metastatico, dell’osso: un follow-up a lungo termine dello studio First Intergroup. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1990; 8: 1664-1674.
- Applebaum MA, et al: Caratteristiche cliniche ed esiti nei pazienti con sarcoma di Ewing e coinvolgimento linfonodale regionale. Pediatric blood & cancer 2012; 59: 617-620.
- Franzius C, et al: FDG-PET per il rilevamento di metastasi polmonari da tumori ossei primari maligni: confronto con la TC spirale. Annali di oncologia: rivista ufficiale della Società Europea di Oncologia Medica/ESMO 2001; 12: 479-486.
- Franzius C, et al: FDG-PET per il rilevamento di metastasi ossee da tumori ossei primari maligni: confronto con la scintigrafia ossea. European journal of nuclear medicine 2000; 27: 1305-1311.
- Salzer-Kuntschik M, et al: Gradi morfologici di regressione nell’osteosarcoma dopo polichemioterapia – studio COSS 80. Journal of cancer research and clinical oncology 1983; 106 Suppl: 21-24.
- Burgert EO Jr, et al: Terapia multimodale per la gestione del sarcoma di Ewing dell’osso non pelvico e localizzato: studio intergruppo IESS-II. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1990; 8: 1514-1524.
- Womer RB, et al: Studio controllato randomizzato sulla chemioterapia a intervalli compressi per il trattamento del sarcoma di Ewing localizzato: un rapporto del Children’s Oncology Group. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2012; 30: 4148-4154.
- Vogin G, et al: Controllo locale e sequele nei tumori di Ewing localizzati della colonna vertebrale: uno studio retrospettivo francese. Eur J Cancer 2013; 49: 1314-1323.
- Puri A, et al: Risultati della resezione chirurgica nel sarcoma di Ewing pelvico. Giornale di oncologia chirurgica 2012; 106: 417-422.
- La TH, et al: Radioterapia per il sarcoma di Ewing: risultati del Memorial Sloan-Kettering nell’era moderna. International journal of radiation oncology, biology, physics 2006; 64: 544-550.
- Krasin MJ, et al: Irradiazione definitiva nella gestione multidisciplinare dei tumori localizzati della famiglia del sarcoma di Ewing nei pazienti pediatrici: esito e fattori prognostici. International journal of radiation oncology, biology, physics 2004; 60: 830-838.
- Grier HE, et al: Aggiunta di ifosfamide ed etoposide alla chemioterapia standard per il sarcoma di Ewing e il tumore neuroectodermico primitivo dell’osso. The New England journal of medicine 2003; 348: 694-701.
- Le Deley MC, et al: Ciclofosfamide rispetto all’ifosfamide nel trattamento di consolidamento del sarcoma di Ewing a rischio standard: risultati dello studio randomizzato di non inferiorità Euro-EWING99-R1. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2014; 32: 2440-2448.
- Whelan J, et al: Efficacia del consolidamento della chemioterapia ad alte dosi busulfano-melfano (BuMel) nel sarcoma di Ewing (ES) localizzato ad alto rischio: risultati dello studio randomizzato EURO-EWING 99-R2 (EE99R2Loc). Journal of Clinical Oncology 2016; 34(15) Suppl: 11000-11000.
- Paulussen M, et al: Tumori di Ewing con metastasi polmonari primarie: analisi della sopravvivenza di 114 pazienti del Cooperative Ewing’s Sarcoma Studies (European Intergroup). Journal of clinical oncology: official journal of the American Society of Clinical Oncology 1998; 16: 3044-3052.
- Letourneau PA, et al: La resezione delle metastasi polmonari nei pazienti pediatrici con sarcoma di Ewing migliora la sopravvivenza. Giornale di chirurgia pediatrica 2011; 46: 332-335.
- Ladenstein R, et al: Sarcoma di Ewing multifocale primario disseminato: risultati dello studio Euro-EWING 99. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2010; 28: 3284-3291.
- Dunst J, Paulussen M, Jurgens H: Irradiazione polmonare per il sarcoma di Ewing con metastasi polmonari alla diagnosi: risultati degli studi CESS. Radioterapia e Oncologia: Organo della Società tedesca Rontgen [et al] 1993; 169: 621-623.
- Paulussen M, et al.: Tumore di Ewing primario metastatico (stadio IV): analisi della sopravvivenza di 171 pazienti degli studi EICESS. Intergruppo Europeo di Studi Cooperativi sul Sarcoma di Ewing. Annali di oncologia: rivista ufficiale della Società Europea di Oncologia Medica/ESMO 1998; 9: 275-281.
- Oberlin O, et al.: Impatto del busulfano ad alte dosi più melfalan come consolidamento nei tumori di Ewing metastatici: uno studio della Societe Francaise des Cancers de l’Enfant. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 2006; 24: 3997-4002.
- Ladenstein R, et al: Impatto della megaterapia nei bambini con tumori di Ewing ad alto rischio in remissione completa: un rapporto del Registro Tumori Solidi EBMT. Trapianto di midollo osseo 1995; 15: 697-705.
- Dirksen U, et al.: Efficacia del consolidamento della chemioterapia ad alte dosi con busulfano-melfalan (BuMel) rispetto alla chemioterapia convenzionale combinata con l’irradiazione polmonare nel sarcoma di Ewing (ES) con metastasi polmonari primarie: Risultati dello studio randomizzato EURO-EWING 99-R2pulm (EE99R2pul). Journal of Clinical Oncology 2016; 34(15) Suppl: 11001-11001.
- Weston CL, et al: Stabilire la sopravvivenza a lungo termine e la cura nei pazienti giovani con sarcoma di Ewing. British journal of cancer 2004; 91: 225-232.
- Stahl M, et al: Rischio di recidiva e sopravvivenza dopo la ricaduta nei pazienti con sarcoma di Ewing. Pediatric blood & cancer 2011; 57: 549-553.
- Bacci G, et al: Resezione di metastasi polmonari metacrone in pazienti con sarcoma di Ewing trattati inizialmente con chemioterapia adiuvante o neoadiuvante. Eur J Cancer 1995; 31A: 999-1001.
- Rasper M, et al: Il valore della chemioterapia ad alto dosaggio nei pazienti con sarcoma di Ewing recidivato per la prima volta. Pediatric blood & cancer 2014; 61: 1382-1386.
- Hunold A, et al: Topotecan e ciclofosfamide nei pazienti con tumori di Ewing refrattari o recidivi. Pediatric blood & cancer 2006; 47: 795-800.
- van Maldegem AM, et al: Terapia combinata di etoposide e carbo-o cisplatino nel sarcoma di Ewing refrattario o recidivato: un ampio studio retrospettivo. Pediatric blood & cancer 2015; 62: 40-44.
- Casey DA, et al: Irinotecan e temozolomide per il sarcoma di Ewing: l’esperienza del Memorial Sloan-Kettering. Pediatric blood & cancer 2009; 53: 1029-1034.
- Ginsberg JP, et al: Sopravvissuti a lungo termine al sarcoma di Ewing infantile: rapporto dello studio sui sopravvissuti al cancro infantile. Journal of the National Cancer Institute 2010; 102: 1272-1283.
InFo ONCOLOGIA & EMATOLOGIA 2018; 6(5): 13-16.